Effect of Nylon-6 Concentration on the Properties of Hot Melt Adhesive Synthesized using Dimer Acid and Ethylenediamine
Pravin G. Kadam and Shashank T. Mhaske*
Department of Polymer and Surface Engineering, Institute of Chemical Technology (formerly UDCT), Matunga, Mumbai - 400 019, India
DOI : http://dx.doi.org/10.13005/msri/090206
Article Publishing History
Article Received on : 12 Oct 2012
Article Accepted on : 20 Nov 2012
Article Published :
Plagiarism Check: No
Article Metrics
ABSTRACT:
Hot melt adhesive synthesized using polymerized fatty acid (PFA) (composition: ~1% trilinoleic acid, ~97% dilinoleic acid and ~2% linoleic acid) and ethylenediamine was blended with nylon-6, in-situ during the synthesis process to improve its performance properties. Nylon-6 was added in concentrations as 5, 10, 15 and 20 phr in the hot melt adhesive. The prepared blends were characterized for thermal (melting temperature, crystallization temperature, enthalpy of melting and enthalpy of crystallization), mechanical (tensile strength, tensile modulus, stiffness, percentage elongation at break and hardness), adhesion (lap shear strength and T-peel strength) and rheological properties. It was found that the viscosity, tensile strength, tensile modulus,
stiffness, hardness, melting temperature, enthalpy of melting, crystallization temperature and enthalpy of crystallization increased with increase in concentration of nylon-6 in the hot melt adhesive. But lap shear strength and T-peel strength increased up to 10 phr concentration of nylon-6 above which both started decreasing. Percentage elongation at break decreased with increase in concentration of nylon-6 in the hot melt adhesive. Hot melt adhesive molecules must have oriented themselves about nylon-6, increasing its crystallinity, and thus the strength of the adhesive.
KEYWORDS:
Polymerized fatty acid; Nylon-6; Hot melt adhesive; Blend; Crystallinity
Copy the following to cite this article:
Kadam P. G, Mhaske S. T. Effect of Nylon-6 Concentration on the Properties of Hot Melt Adhesive Synthesized using Dimer Acid and Ethylenediamine. Mat.Sci.Res.India;9(2)
|
Copy the following to cite this URL:
Kadam P. G, Mhaske S. T. Effect of Nylon-6 Concentration on the Properties of Hot Melt Adhesive Synthesized using Dimer Acid and Ethylenediamine. Mat.Sci.Res.India;9(2). Available from: http://www.materialsciencejournal.org/?p=1114
|
Introduction
Hot melt adhesives (HMA) are solid adhesives that are heated to a molten liquid statefor application to substrates and then cooled, quickly setting up the bond.1 The hot melt systems must achieve a relatively low viscosity when in the molten state to achieve proper wetting of the substrates. However, they must not cool too rapidly as they will not get enough time to completely wet the roughness of the substrates surface. On the application of HMA in the molten condition, the substrates must be joined immediately. HMAs has an open time from a few seconds to few minutes.2 Conventional HMAs cool to harden and do not chemically cross-link.1 An important advantage of hot melts over other adhesive systems is the possibility of being able to pre-apply, e.g. as powder or adhesive spheres, in melt liquid form, as dispersion or as an adhesive foil. The joining procedure does not have to take place directly after applying the adhesive to the substrate; this can happen at any time later on.3-4 The market for HMA is mainly in the manufacture of durable goods such as shoe assembly, kitchen and bathroom cabinets, telecommunication cable repair sleeves, and window assembly.5 HMA are usually clear, off-white, white, or amber coloured compounds. Frequently, photoinitiators are added when UV or other radiation curing is required. Other useful additives include ûllers and reinforcing agents. When there is some lack of cohesiveness in blends of base resins, compatibilizers may be used to improve the apparent miscibility of these resins6. During the cooling process, nucleating agents can also be added to the formulations, to control the development of crystallinity.7
Drawert et al. synthesized flexible HMA using PFA (composition: 2% linoleic acid, 97% dilinoleic acid and 1% trilinoleic acid), ethylenediamine and heptadecane dicarboxylic acid. 70 wt % of total acids used in the system comprised of PFA and remaining 30 wt % of 1,9- heptadecane dicarboxylic acid. Acids and amine were reacted in 1:1 ratio. The prepared HMA gave an extension of about 550 %; whereas softening point was around 106°C and tensile strength was 10.4 MPa.8 Leoni et al. synthesized HMA using PFA (composition: 2% linoleic acid, 97% dilinoleic acid and 1% trilinoleic acid), ethylenediamine and propylenediamine. PFA was reacted with 50 mole % ethylenediamine and 50 mole % propylenediamine. The prepared HMA had the softening point, tensile strength and T-peel strength as 100°C, 8.5 MPa and 1.2 N/mm respectively.9 Rossini et al. studied the synthesis of HMAs using PFA containing 15% trilinoleic acid, 82% dilinoleic acid and 3% linoleic acid. Formulation contained 44 mole% PFA, 6 mole% adipic acid, 30.75 mole% ethylenediamine and 19.25 mole% piperazine. Acids and amines were reacted in 1:1 molar ratio. The prepared HMA had the softening point, T-peel strength and tensile strength of 101°C, 0.72 N/mm and 3.7 MPa respectively10. Kadam et al. studied the synthesis of HMAs using PFA (composition: ~23 % trilinoleic acid, ~75 % dilinoleic acid and ~3 % linoleic acid), sebacic acid, ethylenediamine and piperazine. They found that, as the mole percentage of piperazine or PFA in the polyamide increased, it became more amorphous, decreasing Tf, Hf, Tc, Hc, Ts, Tg, tensile strength, hardness, LSS, TPS and viscosity.11Flanagan et al. synthesized HMAs providing improved adhesion under conditions of high humidity or moisture, by blending them with the silane compounds. HMAs were made up of polyamide using PFA (97% dilinoleic acid) and ethylenediamine. Labels bonded to glass substrate with the above HMA were found to sustain upto 72 hrs; whereas the labels bonded to glass substrate without silane were found to sustain just 24 hrs in humidity resistance test12. Lopez et al. synthesized HMAs by blending acidic ethylene copolymer with PFA. Acidic ethylene copolymer used was ethylenevinyl acetate (23 %)/ methacrylic acid (1%) terpolymer. It was blended in 40:60 ratio with the HMA derived using PFA (97 % dilinoleic acid) and ethylenediamine. The adhesive was found to be suitable for polyethylene and lead bonding applications. Peel strength of polyethylene jacketed cable was 0.7 N/mm while that of lead jacketed cable was 0.53 N/mm.13
Inorder to improve the performance properties of HMA synthesized using PFA (composition: ~1% trilinoleic acid, ~97% dilinoleic acid and ~2% linoleic acid) and ethylenediamine, nylon-6 was blended in situ during the synthesis process. Concentration of nylon-6 was varied up to 20 phr in the HMA. Investigations were made, on
the effect of nylon-6 concentrations on the thermal, mechanical, adhesion and rheological properties of the HMA.
Material and Methods
Materials
Arizona Chemicals, Mumbai, India, supplied dimer acid (Unidyme 18 containing linoleic acid ~ 1%, dilinoleic acid ~ 98%, trilinoleic acid ~1%) with the acid value of 198 mg potassium hydroxide (KOH)/g. Ethylenediamine was procured from Ankita Chemicals, Mumbai, India. Nylon-6 (NXH-01-NC) was obtained from Next Polymers, Faridabad, India. Solvents and indicators were procured from S.D. Fine Chemicals Pvt. Ltd., Mumbai, India. All chemicals were used as obtained without any purification or modification.
Methods
Unidyme 18 consists of 98% dilinoleic acid, 1% trilinoleic acid and 1% linoleic acid. Linoloeic acid and trilinoleic acid were present in very negligible amount, so they were neglected; and Unidyme 18 was supposed to consist of 100% of dilinoleic acid (Dimer acid). Basis for the reaction was 200 gm of Unidyme 18 (200 gm of dilinoleic acid); i.e. it had 0.36 moles of dilinoleic acid. In order to obtain high molecular weight polyamide, the molar ratio of acid to amine was kept at 1. Ethylenediamine, thus, used was 0.36 moles, which is 21.6 g.
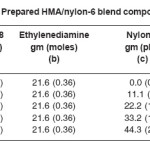
Four necked 500 ml reactor, equipped with stirrer, condenser, nitrogen gas inlet and temperature controller, was loaded with 200 g of Unidyme 18 (0.36 mol dilinoleic acid), 21.6 g (0.36 mol) of ethylenediamine and the calculated amount of nylon-6 (phr), under continuous stirring at 1500 rpm. The mixture was heated gradually upto 250°C with continuous stirring. Product was removed from the reactor when the acid value of the blend system was obtained to be below 10 mg KOH/gm sample. Time required to reach the desired acid value was 4-5 hours. Prepared blend system was then poured in a teflon mold (having dimension: 15×15×3 cm3), in the molten condition, and was allowed to get cooled at room temperature. Prepared hot melt/ nylon-6 blend compositions are shown in the Table 1.
Differential scanning calorimetry (DSC Q100, TA Instruments, USA) was used to investigate the melting and crystallization behaviour of the prepared blend systems. Two consecutive heating scans were obtained to minimize the inûuence of possible residual stresses in the material due to any speciûc thermal history. A scanning rate of 10°C/ minute was used for both heating and cooling cycles with the nitrogen purge rate maintained at 50 mL/ minute. The enthalpy of melting (Hm) was determined from the second heating cycle, whereas the enthalpy of crystallization (Hc) was determined from first cooling scan. The area under the melting or crystallization peak represents the amount of energy required to melt the polymer (Hm) and the amount of energy released during crystallization (Hc) respectively. The peak area calculation is used with the limits of the calculation on the flat portion of the baseline before and after the melting or crystallization peak. Peak temperatures were noted as the melting (Tm) and crystallization (Tc) temperatures.
The FTIR spectra were recorded with Perkin Elmer, Spectrum GX equipment. 2-4 gm of blend was dissolved in chloroform for the characterization and was scanned with a resolution of 2 cm-1 in the scan range of 450–4000 cm-1. FTIR of pure solvent was run prior to running the FTIR of the samples, to use its peaks as baseline, so as to automatically subtract it from the samples peak.
Tensile strength of compression molded sheet was measured in accordance to ASTM D638 at a crosshead speed of 50 mm/min. Shore D hardness was determined in accordance with ASTM D2240. Polyamide adhesive films were obtained by compression molding. The adhesive joint was then prepared by pressing the film between two pieces of surface treated aluminium substrate at a temperature of 200°C and at a pressure of 30 KPa for 20 min, followed by cooling at room temperature for 24 h. LSS was determined according to ASTM D1002-72 at a crosshead speed 1.3 mm/min. TPS was measured in accordance to ASTM D1876 at a crosshead speed of 254 mm/ min. Average thickness of adhesive layer after formation of the lap and T-peel joints were kept within 0.035 mm (0.001 inch). Viscosity vs shear rate was determined at the temperature of 220°C up to a maximum shear rate of 100 s-1. Viscosity vs time was determined at temperature of 220°C at constant shear rate of 1 s-1 for 200 seconds. Viscosity was determined using rheometer (MCR101, Anton Paar, Germany).
Results and Discussion
Acid and Amine Value
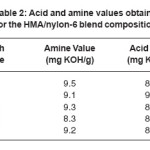
The acid and amine values determined for the blends are listed in Table 2. Since acid value and amine value did not vary significantly, it was supposed that the number of acid and amine functional groups in all the blend systems are approximately same. Thus, it will be the concentration of nylon-6 in the HMA, which would affect the mechanical, thermal, adhesion and rheological properties and not the acid or amine end groups of the HMA.
Table 1: Prepared HMA/nylon-6 blend compositions
FTIR Analysis
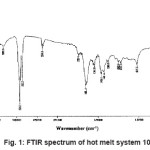
FTIR spectrum of the blend system 10N is shown in the Figure 1. Small band of the amide I (– NH2 in primary amides) groups appeared at 3441.9 cm-1 and 3320.8 cm-1. Peaks at 2924.4 cm-1 and 2853.5 cm-1 were due to asymmetric and symmetric stretching of CH2, respectively. The carbonyl peak (–CONH2) of polyamide was at around 1651.4 cm-1 (steep peak). Amide II band/CH2 asymmetric deformation was indicated by the peak at wave number 1542.2 cm-1. N-H deformation/CH2 scissoring peak was indicated at wave number 1456.0 cm-1. Peak at 1369.0 cm-1 was due to amide III band/CH2 wagging. The peak at 1243.9 cm-1 was corresponded to C–N stretching vibration bond. Small peak at 773.6 cm-1 showed C-C deformation. Similar method and same raw materials were used for synthesizing other systems (UE to 20 N), thus, they are also amides.
Table 2: Acid and amine values obtained for the HMA/nylon-6 blend compositions
Thermal Analysis
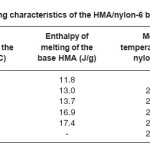
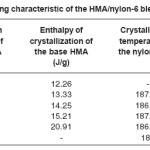
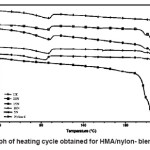
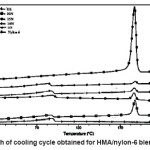
Heating and cooling curves of the blend systems are shown in Figures 2 and 3 respectively. Values of Tm, Tc, Hm and Hc obtained for the blend systems are listed in the Tables 3 and 4. First heating and crystallization peaks in the graph belong to the hot melt adhesive synthesized using unidyme 18 (dimer acid) and ethylenediamine, whereas the second peaks belong to the nylon-6. It was observed that as the concentration of nylon-6 increased in the HMA, melting temperature and crystallization temperature of the HMA increased whereas that of nylon-6 decreased. This is clear indication of better interaction and dispersion of nylon-6 in HMA. Due to better dispersion, HMA molecules got better oriented about nylon-6, increasing the crystallinty of HMA, thus increasing its melting and crystallization temperature. At the same time, as nylon-6 got better dispersed in HMA, the intermolecular forces of attraction between nylon-6 molecules decreased, decreasing the melting and crystallization temperature. However, enthalpy of melting and enthalpy of crystallization of both the base HMA and the added nylon-6 increased with increase in nylon-6 concentration. As the concentration of nylon-6 in the base HMA increased, the number of nylon-6 polymeric chains increased requiring more heat for melting, increasing enthalpy of melting and would also release more heat during cooling, increasing the enthalpy of crystallization.
Table 3: Heating Characteristics of the HMA/Nylon-6 Blends Prepared
Interestingly, the enthalpy of melting and crystallization also increased for the base HMA, which happened due to better interaction between nylon-6 and base HMA, requiring more heat to break the interactions.
Similar acid value and amine value suggests that same number of amide linkages must have been formed in all the base HMAs of the blend systems. Nylon-6 and the base HMA are both polyamides, having better compatibility. Nylon-6 is more crystalline than the base HMA, and also have higher molecular weight. Thus, it may have acted as a reinforcing agent in the base polymer. Small molecules of base HMA might have oriented themselves over the molecules of nylon-6 increasing the crystallinity of the system, as evident from the enthalpy of melting of the base HMA. Mechanical properties:
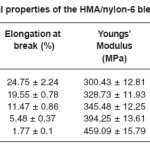
Table 5 lists the tensile strength, percentage elongation at break, stiffness, tensile modulus and shore D hardness values obtained for the blend systems. It was observed that the tensile strength, Youngs modulus, stiffness and Shore D hardness increased with increase in the concentration of nylon-6 in the HMA.
Table 4: Cooling Characteristic of the HMA/Nylon-6 Blends Prepared
Table 5: Mechanical Properties of the HMA/Nylon-6 Blend Systems Prepared
Table 6: Adhesion properties of the HMA/nylon-6 blend systems prepared
In composites, it is a known fact that the addition of any reinforcing agent improves the crystallinity of the material increasing its stiffness, modulus, whereas decreasing the elongational property. Similar trend was observed for the addition of nylon-6 on the properties of the base HMA. Thus, nylon-6 induces crystallinity into the base HMA. This must have happened due to better interaction between the base HMA and nylon-6, which was due to better compatibility between the two. As the concentration of nylon-6 in the base HMA increased more amide linkages became available, increasing hydrogen bonding between nylon-6 and hot melt polymer. Small molecules of base HMA must have oriented themselves over the better arranged molecules of nylon-6 increasing the crystallinity of the system. This proves the role of nylon-6 as reinforcing agent in the HMA. Tensile strength also increased with increase in nylon-6 in the hot melt. This is due to the better compatibility between the two. Nylon-6 holds the hot melt molecules together, resisting their movement to the applied tensile force, increasing tensile strength.
This resistance to polyamide molecular chains leads to increase in crystallinity of the system, thus decreasing the elongational property.
Tensile strength, stiffness, tensile modulus and shore D hardness increased by 37.3, 39.26, 52.8 and 25% respectively at 20 phr loaded samples. Percentage elongation at break was found to have decreased by 92%.
Adhesion properties
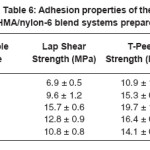
Table 6 lists the T-peel strength (TPS) and lap shear strength (LSS) values obtained for the blend systems. The TPS and LSS increased with increase in concentration of nylon-6, until its concentration reached 10 phr, above which both the strengths started decreasing.
Figure 1: FTIR Spectrum of Hot Melt System 10N
Figure 2: Graph of Heating Cycle Obtained for HMA/Nylon- Blend Systems
Nylon-6 increased the crystallinity of the system due to the proper orientation and hydrogen bonding induced by it onto the hot melt polymer. This was mainly due to better interaction between the two polymers caused by amide linkages. This interaction increased with increase in nylon-6 concentration in the base HMA. This molecular orientation and interaction proved helpful until the concentration of nylon-6 reached 10 phr. As till then, there was better interaction of the blend system
with the substrate, due to proper wetting of the substrate. But as the concentration of nylon-6 exceeded 10 phr, crystallinity as well as viscosity (see the rheology section below) of the blend system increased so much, that they were not able to have better interaction with the substrate (improper wetting); thus resulting in interfacial failure, leading to decrease in LSS and TPS of the system.
LSS and TPS were found to have increased by 127.5 and 80.7% respectively at 10 phr loading of nylon-6 in the HMA, which is highly appreciable compared to the amount of nylon-6 added. This is beneficial not only from the performance point of view but also from the cost point.
Figure 3: Graph of Cooling Cycle Obtained for HMA/Nylon-6 Blend Systems
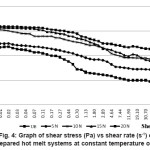
Figure 4: Graph of Shear Stress (Pa) vs Shear Rate (s-1) of the Prepared Hot Melt Systems at Constant Temperature of 225°C
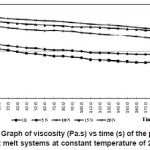
Figure 5: Graph of Viscosity (Pa.s) Vs Time (s) of the Prepared Hot Melt Systems at Constant Temperature of 225°C
Rheological Properties
The viscosity vs shear rate of the blend systems investigated at the constant temperature of 225°C is shown in Figure 4. Viscosity of the system increased with increase in nylon-6 concentration. Intermolecular forces of attraction increased with increase in nylon-6 concentration, as evident from mechanical properties, resisting the shearing forces exerted by the rotating spindle of the rheometer. Zero shear viscosity also increased with increase in the nylon-6. Blend system showed shear thinning behaviour. Viscosity decreased with increase in shear rate proving the pseudo-plastic nature of the material. The rate of decrease of viscosity with shear rate increased with increase in concentration of nylon-6, indicating the presence of nylon-6, which is a material of very low melt viscosity. But, still the viscosity increased with increase in nylon-6, due to the increase in intermolecular forces of attraction between base HMA and the nylon-6.
To understand how time had an effect on viscosity, study of viscosity vs time was carried out at constant temperature of 225°C and constant shear rate of 1 s-1 and is shown in the Figure 5. Blend systems showed thixotropic behaviour. This can be due to breaking and forming of the molecular aggregate. But, in general, the viscosity increased with increase in nylon-6 concentration, due to the increased intermolecular forces of attraction. Appreciable increase in viscosity w.r.t. can be seen
with the addition of nylon-6.
References
- Petrie E.M., Handbook of Adhesives and Sealants, McGraw-Hill Professional, New York, 135-139 (2000).
- Yorkgitis E.M., Encyclopedia of Polymer Science and Technology Adhesive Compounds, John Wiley & Sons Inc., New York, 256-270 (2001).
- Cagle C.V., Handbook of Adhesive bonding, McGraw-Hill Professional, New York, 334- 389 (1982).
- Böhm S., Hemken G., Stammen E. and Dilger K., J. Adhes. Interfac., 7: 28 (2006).
- Frihart C.R., Inter. J. Adhes. Adhes., 24, 415 (2004).
- Piglowski J., Trelinska-Wlazlak M. and Paszak B., J. Macromol. Sci. Part B., 38: 515 (2006).
- Ghosh S., Khastgir D., and Bhowmick A.K., J. Adhes. Sci. Tech., 14: 529 (2000).
- Drawert M., Griebsch E. and Schering A.G., US Patent No. 3: 781,234 (1973).
- Leoni R., Gruber W. and Wichelhaus J., US Patent No. 4, 914: 162 (1990).
- Rossini A. and Meda F., US Patent No. 6, 670: 442 (2003).
- Kadam P.G. and Mhaske S.T., Inter. J. Adhes. Adhes., 31: 735 (2011).
- Flanagan T.P. and Kaye I., US Patent No. 3, 644: 245 (1972).
- Lopez E.F., Glover L.C. and Lyons B.J., US Patent No. 4,018,733 (1977).

This work is licensed under a Creative Commons Attribution 4.0 International License.