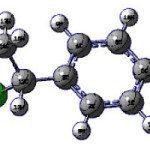
IR, Raman, First Hyperpolarizability and Computational Study of 1-chloroethyl Benzene
Hema Tresa Varghese1, C. Yohannan Panicker2* and Sheena Mary Y1
¹Department of Physics, Fatima Mata National College, Kollam, Kerala, India.
²Department of Physics, TKM College of Arts and Science, Kollam, Kerala, India.
DOI : http://dx.doi.org/10.13005/msri/090116
Article Publishing History
Article Received on : 03 Feb 2012
Article Accepted on : 19 Mar 2012
Article Published :
Plagiarism Check: No
Article Metrics
ABSTRACT:
The FTIR and FT-Raman spectra of 1-chloroethylbenzene were recorded and analyzed. The harmonic vibrational wavaenumbers were calculated theoretically using Gaussian03 set of quantum chemistry codes. The calculated wavenumbers (B3LYP) agree well with the observed wavenumbers. The calculated first hyperpolarizability is reported and the title compound is an attractive object for further studies of nonlinear optics.
KEYWORDS:
Chloroethyl; DFT; FT-IR; FT-Raman; Hyperpolarizability
Copy the following to cite this article:
Varghese H. T, Panicker C. Y, Mary Y S. , IR, Raman, First Hyperpolarizability and Computational Study of 1-chloroethyl Benzene. Mat.Sci.Res.India;9(1)
|
Copy the following to cite this URL:
Varghese H. T, Panicker C. Y, Mary Y S. , IR, Raman, First Hyperpolarizability and Computational Study of 1-chloroethyl Benzene. Mat.Sci.Res.India;9(1). Available from: http://www.materialsciencejournal.org/?p=1201
|
Introduction
Chloroethyl derivatives are new agents that have shown promising cytotoxic and antineplastic activities. The cytotoxicity of many chloroethyl derivatives was demonstrated on several tumor cell lines.1 Kulkarni et al.,2 reported the quantitative structure activity relationship studies of chloroethyl derivatives. Nonlinear optics deals with the interaction of applied electromagnetic fields in various materials to generate new electromagnetic fields, altered in wavenumber, phase, or other physical properties.3 Organic molecules able to manipulate photonic signals efficiently are of importance in technologies such as optical communication, optical computing, and dynamic image processing.4,5 In this context, the dynamic first hyperpolarizability of the title compound is also calculated in the present study. The first hyperpolarizability (b0) of this novel molecular system is calculated using B3LYP/6- 31G(d) method, based on the finite field approach. In the presence of an applied electric field, the energy of a system is a function of the electric field. First hyperpolarizability is a third rank tensor that can be described by a 3 × 3 × 3 matrix. The 27 components of the 3D matrix can be reduced to 10 components due to the Kleinman symmetry.6 The components of b are defined as the coefficients in the Taylor series expansion of the energy in the external electric field. When the electric field is weak and homogeneous, this expansion becomes
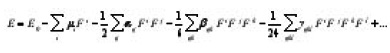
where Eo is the energy of the unperturbed molecule, Ei is the field at the origin, μi, αij, βijk and Yijk are the components of dipole moment, polarizability, the first hyper polarizabilities, and second hyperpolarizibilites, respectively. In the present work, vibrational spectroscopic analysis of 1-chloroethyl benzene is reported.
Experimental
The FT-IR spectrum was recorded on a DR/Jasco FT-IR 6300 spectrometer and the FT- Raman spectrum was obtained on a BRUKER RFS 100/S, Germany. For excitation of the spectrum the emission of a Nd:YAG laser was used, excitation wavelength 1064 nm, maximal power 150 mW. One thousand scans were accumulated with a total registration time of about 30 min. The spectral resolution after apodization was 2 cm-1.
Computational Details
Calculations of the title compound were carried out with Gaussian03 software program7 using the B3LYP/6-31G* basis sets to predict the molecular structure and vibrational wavenumbers. The DFT hybrid B3LYP functional tends also to overestimate the fundamental modes; therefore scaling factors have to be used for obtaining a considerably better agreement with experimental data. Therefore, a scaling factor of 0.9613 was uniformly applied to the DFT calculated wavenumbers.8
Scheme 1
Results and Discussion
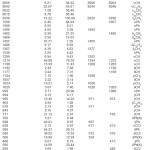
The observed IR, Raman and calculated (scaled) wavenumbers and the assignments are given in Table 1. The phenyl CH stretching modes occurs above 3000 cm-1 and is typically exhibited as multiplicity of weak to moderate bands compared with the aliphatic CH stretching.9 In the present case, the DFT calculations give υCH modes of the phenyl ring at 3103, 3093, 3084, 3074 and 3066 cm-1. The bands observed at 3090, 3068 cm-1 in the IR spectrum and at 3064 cm-1 in the Raman spectrum were assigned as CH stretching modes of the phenyl ring. The benzene ring possesses six ring stretching vibrations, of which the four with the highest wavenumbers (occurring near 1600, 1580,1490 and 1440 cm-1) are good group vibrations.10 With heavy substituents, the bands tend to shift to somewhat lower wavenumbers. In the absence of ring conjugation, the band at 1580 cm-1 is usually weaker than that at 1600 cm-1. In the case of C=O substitution, the band near 1490 cm-1 can be very weak. The fifth ring stretching vibration is active near 1315 ± 65 cm-1, a region that overlaps strongly with that of the CH in-plane deformation.10 The sixth ring stretching vibration, or the ring breathing mode, appears as a weak band near 1000 cm-1, in mono, 1,3-di- and 1,3,5-trisubstituted benzenes. In the otherwise substituted benzenes, however, this vibration is substituent sensitive and difficult to distinguish from the ring in-plane deformation.10,11 For mono substituted benzene, the õPh modes are expected in the range10 of 1285-1610 cm-1. For the title compound, the phenyl ring stretching modes are assigned at 1494, 1455 cm-1 in the IR spectrum, 1607 cm-1 in the Raman spectrum and at 1598, 1580, 1496, 1457, 1330 cm-1 theoretically. The ring breathing mode is observed at 1006 cm-1 in the Raman spectrum which finds support from the computational value at 999 cm-1, as expected.
The in-plane CH vibrations are expected in the range of 1015-1300 cm-1 for mono substituted benzenes, respectively.10 For the title compound, the DFT calculations give 1299, 1192, 1177, 1057, 1025 cm-1 as nCH modes. These modes are observed at 1303, 1028 cm-1 in the IR spectrum and at 1190, 1168, 1029 cm-1 in the Raman spectrum. The out-of-plane CH deformations10 are observed between 1000 and 700 cm-1. Generally, the CH out-of-plane deformations with the highest wavenumbers have a weaker intensity than those absorbing at lower wavenumbers. These ãCH modes are observed at 971, 911 cm-1 in the IR spectrum and at 972, 782 cm-1 in the Raman spectrum. The DFT calculations give these modes at 994, 966, 918, 843 and 771 cm-1. In the present case, the out-of-plane CH deformation at 782 cm-1 and the ring deformation at 697 cm-1 in the IR spectrum form a pair of strong bands characteristics of mono substituted benzene.12
In aromatic compounds the asymmetric stretching vibrations of CH3 are expected in the range 2905-3000 cm-1 and symmetric CH3 vibrations in the range10,13 of 2860-2900 cm-1. The first of this results from the asymmetric stretching νasCH3 mode in which two C-H bonds of the methyl group are extending while the third one is contracting. The second arises from the symmetrical stretching νsCH3 in which all three of the C-H bonds extend and contract in phase. The asymmetric stretching modes of the methyl group are calculated (DFT) to be 3022, 3010 cm-1 and the symmetric mode at 2936 cm-1. The bands observed at 2929 cm-1 in the IR spectrum and 2930 cm-1 in the Raman spectrum are assigned as stretching modes of the methyl group.Two bending can occur within a methyl group. The first of these, the symmetrical bending vibration, involves the in-phase bending of the C-H bonds. The second, the asymmetrical bending vibration, involves out-of-phase bending of the C- H bonds. The asymmetrical deformations10 are expected in the range 1400-1485 cm-1. The calculated values (DFT) of δasCH3 modes are at 1482, 1468 cm-1. In many molecules, the symmetric deformations δsCH3 appears with an intensity varying from medium to strong and expected in the range10 1380 ± 25 cm-1. The band observed at 1377 cm-1 in the IR spectrum is assigned as the δsCH3 mode. The DFT calculations give δsCH3 mode at 1367 cm-1. Aromatic molecules display10 a methyl rock in the neighborhood 1045 cm-1. The second rock10 in the region 970±70 cm-1 is more difficult to find among the C-H out-of-plane deformations. In the present case, these νCH3 modes are calculated at 1104 and 1053 cm-1. The band observed at 1090, 1050 cm-1 in the IR spectrum assigned as rocking modes of the methyl group. The methyl torsions often assigned in the region10 185 ± 65 cm-1.
The aliphatic CCl bands absorb13 at 830- 560 cm-1 and putting more than one chlorine on a carbon atom raises the CCl wavenumber. The CCl2 stretching mode is reported11,13 at around 738 cm-1 for dichloromethane and scissoring mode δCCl2 at around 284 cm-1. Pazdera et al.14,15 reported the CCl stretching mode at 890 cm-1. For 2- cyanophenylisocyanide dichloride, the νCCl stretching mode is reported at 870 cm-1 (IR), 877 cm-1 (Raman) and at 882 cm-1 theoretically.16 Arslan et al.17 reported νCCl at 683 (experimental) and at 711, 736, 687, 697 cm-1 theoretically. The deformation bands of CCl are reported at 431, 435, 441 and 443 cm-1. In the present case, the band at 617 (IR), 619 (Raman) and 628 cm-1 (DFT) is assigned as the stretching mode of CCl. The deformation bands of the CCl are assigned below 400 cm-1. The C-C stretching modes are assigned at 1214, 1198 (DFT), 1234, 1200 (IR) and 1225, 1203 cm-1 (Raman) as expected10. The substituent sensitive modes of the phenyl ring is also identified and assigned (Table 1).
Using the x, y and z components, the magnitude of the dynamic first hyperpolarizability can be calculated by
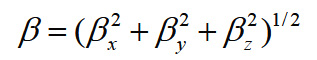
The complete equation for calculating the magnitude of the dynamic first hyperpolarizability from the Gaussian03 output is given as follows.18
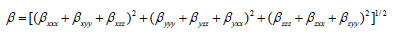
To calculate the dynamic first hyperpolarizability, the origin of the Cartesian coordinate system was chosen as the centre of mass of the compound. The calculated first hyperpolarizability of the title compound in an electric field of value 0.001 au is 1.56 × 10-30 esu. We conclude that the title compound is an attractive object for future studies of non linear optical properties.
Table 1: Vibrational assignments of 1-chloroethyl benzene
Conclusion
The FTIR and FT-Raman spectra of 1- chloroethylbenzene were recorded and analyzed. The harmonic vibrational wavaenumbers were calculated theoretically using Gaussian03 set of quantum chemistry codes. The calculated wavenumbers (B3LYP) agree well with the observed wavenumbers. The calculated first hyperpolarizability is reported and the title compound is an attractive object for further studies of nonlinear optics.The predicted infrared intensities and Raman activities are reported
References
- Bechard, P., Lacroix, J., Poyet, P., and Gaudreault, R., Eur. J. Med. Chem. 29: 963 (1994).
CrossRef
- Kulkarni, G.B., Reddy, A.S., Padmavathi, A., and Venkateswararao, M., Int. J. ChemTech. Res. 1: 727 (2009).
- Shen, Y.R., The Principles of Nonlinear Optics, Wiley, New York (1984).
- Kolinsky, P.V., Opt. Eng. 31: 1676(1992).
CrossRef
- Eaton, D.F., Science, 253: 281 (1991).
CrossRef
- Kleinman, D.A, Phys. Rev. 126: 1977 (1962).
CrossRef
- Frisch, M.J., et al., Gaussian 03, Revision C.02 Gaussian, Inc., Wallingford CT (2004).
- Foresman, J.B., in: Frisch, E., (Ed.), Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian”, Pittsburg, PA (1996).
- Coates, J., in Encyclopedia of Analytical Chemistry: Interpretation of Infrared spectra, A Practical Approach (Ed. Meyers, R.A.), John Wiley andSons, Chichester (2000).
- Roeges, N.P.G., A Guide to the Complete Interpretation of Infrared Spectra of Organic Structures, Wiley: New York (1994).
- Varsanyi, G., Assignments of vibrational Spectra of Seven Hundred Benzene Derivatives, Wiley, New York (1974).
- Higuchi, S., Tsuyama, H., Tanaka, S., and Kamada, H., Spectrochim Acta, 30: 463 (1974).
CrossRef
- Colthup, N.B., Daly, L.H., and Wiberly, S.E., Introduction to Infrared and Raman Spectroscopy, second ed., Academic Press, New York (1985).
- Pazdera, P., Divisova, H., Havilsova, H., and Borek, P., Molecules 5: 189 (2000).
CrossRef
- Pazdera, P., Divisova, H., Havilsova, H., and Borek, P., Molecules 5: 1166 (2000).
CrossRef
- Varghese, H.T., Panicker, C.Y., Philip, D., and Pazdera, P., Spectrochim.Acta, 67A: 1055 (2007).
- Arslan, H., Florke, U., Kulcu, N., and Binzet, G., Spectrochim. Acta, 68A: 1347 (2007).
- Thanthiriwatte, K.S., and Silva, K.M.N., J. Mol. Struct. Theochem. 617: 169 (2002).
CrossRef



This work is licensed under a Creative Commons Attribution 4.0 International License.