Computational Insights on Molecular Structure, Electronic Properties, and Chemical Reactivity of (E)-3-(4-Chlorophenyl)-1-(2-Hydroxyphenyl)Prop-2-en-1-one
Vishnu A. Adole1 , Prashant B. Koli2*, Rahul A. Shinde1 and Rohit S. Shinde1
, Prashant B. Koli2*, Rahul A. Shinde1 and Rohit S. Shinde1
1Department of Chemistry, Mahatma Gandhi Vidyamandir’s Arts, Science and Commerce College, Manmad, (Affiliated to SP Pune University, Pune) Nashik-423 104, India
2Department of Chemistry, Arts, Commerce and Science College, Nandgaon, (Affiliated to SP Pune University, Pune) Nashik-423 106, India
Corresponding Author Email: prashantkoli005@gmail.com
DOI : http://dx.doi.org/10.13005/msri.17.special-issue1.06
Article Publishing History
Article Received on : 24 April 2020
Article Accepted on : 27 May 2020
Article Published : 29 May 2020
Plagiarism Check: Yes
Reviewed by: Raheleh Farahani
Second Review by: Uday Kiran
Final Approval by: Dr MGH ZAIDI
Article Metrics
ABSTRACT:
In the current examination, (E)-3-(4-chlorophenyl)-1-(2-hydroxyphenyl)prop-2-en-1-one has been studied to investigate geometrical entities, electronic properties, and chemical reactivity viewpoints. To inspect structural, spectroscopic, and chemical reactivity aspects, density functional theory method (DFT) at B3LYP/6-311G(d,p) basis set has been employed. The (E)-3-(4-chlorophenyl)-1-(2-hydroxyphenyl)prop-2-en-1-one has been synthesized and characterized by FT-IR, 1HNMR, and 13C NMR spectral techniques. The detailed investigation of bond lengths and bond angles is discussed to comprehend the geometrical framework. To explore its chemical behaviour, Mulliken atomic charges, molecular electrostatic potential surface, and electronic parameters are introduced. The imperative exploration of the electronic properties, such as HOMO and LUMO energies, was studied by the time-dependent DFT (TD-DFT) method. The dipole moment of the title molecule is 2.57 Debye with C1 point group symmetry. The most electropositive carbon and hydrogen atoms in the title molecule are C14 and H27 respectively. Amongst aromatic C=C, the C16-C18 is the longest, and C17-C19 is the shortest bond. The molecular electrostatic potential plot predicts the positive electrostatic potential is around hydrogen atoms. The vibrational assignments were made by comparing the experimental FT-IR absorption peaks with the scaled frequencies obtained using computational work. Besides, some significant thermochemical information is obtained using the same basis set using frequencies.
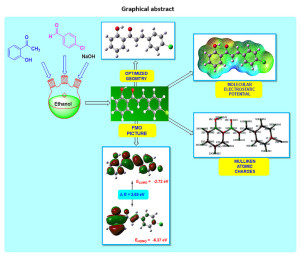
Graphical Abstract
KEYWORDS:
Chalcones; B3LYP/6-311G (d; p); FMO; Molecular Electrostatic Potential
Copy the following to cite this article:
Computational Insights on Molecular Structure, Electronic Properties, and Chemical Reactivity of (E)-3-(4-Chlorophenyl)-1-(2-Hydroxyphenyl)Prop-2-en-1-one. Mat. Sci. Res. India; Special Issue (2020).
|
Copy the following to cite this URL:
Computational Insights on Molecular Structure, Electronic Properties, and Chemical Reactivity of (E)-3-(4-Chlorophenyl)-1-(2-Hydroxyphenyl)Prop-2-en-1-one. Mat. Sci. Res. India; Special Issue (2020). Available from: https://bit.ly/2TNtMKY
|
Introduction
The rising attention towards chalcone derivatives is the direct consequence of their broad pharmacological applications.1 Chalcones are naturally occurring biologically potent compounds belonging to the flavonoid family (Figure 1). Apigenin (1), luteolin (2), chrysoeriol (3), and velutin (4) are some important examples of flavones (Figure 2). Synthetically, chalcones are synthesized by Claisen-Schmidt reaction of aromatic ketones and aromatic aldehydes having 1,3- diphenylprop-2-en-1-one as a basic framework. The fantastic medicinal and pharmacological pattern of chalcones includes properties like anticancer,2 antitumor,3 anti-diabetics,4 anti-HIV,5 antioxidants,6 antiulcer,7 antimalarial,8 cardiovascular,9 anti-tubercular,10 antibacterial,11 antifungal,12 antiviral,13 anti-inflammatory,14 anticonvulsant,15 antidiuretic,5 etc. The 2-arylidene indanone derivatives are having a structural framework similar to the chalcones.16 These compounds have also been explored as biological agents. Besides, chalcones have also been used as versatile intermediates for the synthesis of a variety of heterocyclic compounds having promising therapeutic applications.17-20 The biologically active aza, thia, and oxa synthetic heterocyclic compounds were synthesized from chalcones.21-23 Importantly, the chalcone framework can be synthetically manipulated to get a diverse pattern of biological properties.
Figure 1: Structure of chalcone and flavone
Figure 2: Some examples of flavones
The DFT computations have been used to study various molecular and spectroscopic properties of the molecules. The compounds like nicotinic acid N-oxide,24 sesquiterpene lactone,25 mesitylene,26 5-nitro-2-(4-nitrobenzyl) benzoxazole,27 2-arylidene indanone,28 3,5-difluorophenylboronic acid,29 cyclohexanone oxime,30 2-methylpyridine 1-oxide,31 pycolinaldehydeoxime,32 o-methoxybenzoates,33(E)-1-(4-bromobenzylidene)semicarbazide,34 1,5-diphenylpenta-1,4-dien-3-one,35 3-(2-Chloro-6-fluorophenyl)-1-(2-thienyl) prop-2-en-1-one,36 dihydropyrimidinone,37 6,8-dichloro-2-(4-chlorophenyl)-4H-chromen-4-one,38 of 2-[5-(4-chlorophenyl)-4, 5-dihydro-1H-pyrazol-3-yl] phenol,39 etc. have been studied to explore their structural, electronic, chemical, and spectroscopic facets. In the last decade majority of researchers are working on computational models utilizing some potential nanomaterials for theoretical aspects.40-43 .Utilization of green chemistry principles has been advanced in recent years to reduce the environmental hazard.44 Considering all these crucial a successful attempt has been made to study structural, electronic, chemical, and spectroscopic properties of the (E)-3-(4-chlorophenyl)-1-(2-hydroxyphenyl)prop-2-en-1-one (4CPHPP). To the best of understanding, this is a primary report on the combined theoretical and experimental study of the title compound 4CPHPP by DFT method at B3LYP/6-311G(d,p) basis set.
Experimental Details
General Remarks
The chemicals (Make- SD fine chemicals and Avra synthesis) were purchased from Sigma laboratory, Nashik with a high purity of 99 %, and they were used as such without any purification. The FT-IR spectrum of the title compound was recorded on Shimadzu spectrometer. The sample was prepared using a KBr disc technique. The NMR experiment was performed on sophisticated multinuclear FT NMR Spectrometer model Avance-II (Bruker). The compound was dissolved in DMSO-d6. Chemical shifts were reported in ppm relative to tetramethylsilane (TMS) for 1H NMR spectra. The reaction was monitored by thin-layer chromatography using Merck Aluminum TLC plate, silica gel coated with fluorescent indicator F254. All the glass apparatus were cleaned and dried in an oven before use.
Experimental procedure for the synthesis of the title compound 4CPHPP
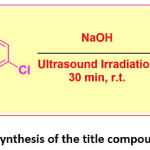
The title compound 4CPHPP was synthesis as per previously reported method.45 In a typical synthesis method 2-hydroxy acetophenone (0.01 mol) and 4-chloro benzaldehyde (0.01 mol) were mixed in ethanol solvent. To this appropriate amount of aqueous NaOH solution was added. Then the alkaline mixture was exposed to ultrasound irradiation. After completion of the reaction (monitored by TLC), the reaction was quenched by pouring onto the crushed ice. It was then acidified, filtered, dried, and recrystallized to give pure crystals of the title compound. The reaction is presented in Scheme 1.
Scheme 1: Synthesis of the title compound 4CPHPP
Computational method
The molecular structure of 4CPHPP in the ground state (in the gas phase) was optimized by DFT/B3LYP method with a 6-311G(d,p) basis set level, and the optimized structure was used in the vibrational frequency calculations. The molecular geometry optimization has been performed by using Gauss View 4.1 software package. The calculated harmonic vibrational frequencies were scaled by 0.96. The entire calculations were implemented by using Gauss View 4.1 program and Gaussian 03W program package on a computing system.46 To explore the electronic properties, the theoretical UV-Visible spectra have been investigated by the TD-DFT method with a 6-311G(d,p) basis in the gas phase.
Results and Discussion
Spectral data of the title compound 4CPHPP
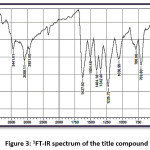
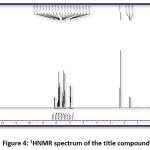
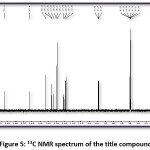
FT-IR (cm-1) – 3441.01, 3086.11, 2931.80, 1627.92, 1504.48, 1404.18, 1342.46, 1226.73, 1056.99, 786.96; 1HNMR (400 MHz, DMSO-d6); δ (ppm) – 6.98 (m, 2H), 7.19 (m, 2H), 7.43 (m, 2H), 7.52 (m, 1H), 7.83 (m, 2H), 7.89 (m, 2H), 8.13 (m, 1H), 12.69 (s, 1H); 13C NMR (400 MHz, DMSO-d6); δ (ppm) – 117.68, 118.76, 120.06, 121.61, 128.70, 130.37, 130.52, 133.11, 135.68, 136.14, 143.28, 162.45, 193.32.
Figure 3: 1FT-IR spectrum of the title compound
Figure 4: 1HNMR spectrum of the title compound
Figure 5: 13C NMR spectrum of the title compound
The title compound 4CPHPP has been successfully characterized by FT-IR (Figure 3), 1H NMR (Figure 4), and 13C NMR (Figure 5), spectroscopic methods. The important FT-IR absorption signals are 3441.01 cm-1 (due to OH stretching vibrations), 3086.11 cm-1(due to Ar-CH stretching vibrations), 1627.92 cm-1 (due to C=O and C=C stretching vibrations), and 1504.18 cm-1(Ar C=C stretching vibrations). The important chemical shift value in 1H NMR spectrum is singlet at 12.69 δ which is assigned to O-H proton. The high deshielding is due to the intra-molecular hydrogen bonding between O-H proton and the carbonyl oxygen atom. This is also confirmed in the computational results. Other signals and the total number of protons are exactly matching with the structure of the title compound. The total carbons in the title compound 4CPHPP are 15; however in the 13C NMR spectrum thirteen signals have appeared. This is due to the symmetric nature of the 4-chlorphenyl ring.
Computational Results
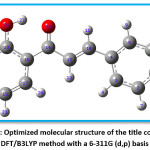
The optimized geometry extracted by using Gauss View 4.1 program and Gaussian 03W program package is depicted in the Figure 6. In the title compound 4CPHPP, there are a total of 29 atoms are present. The structural entities; bond lengths and bond angles are obtained for the optimized structure are presented in Table 1. The important bond length values are C14=O15 (1.2441 Å), C12=C18 (1.3453 Å), and O26-H27 (1.0845 Å). The increase in the bond length of the carbonyl group is due to the presence of intramolecular hydrogen bonding. The C1-Cl29 bond length is 1.7549 Å. The hydrogen bond between the oxygen of carbonyl and hydrogen of hydroxyl has bond length 1.6372 Å. The presence of this bond in the computational result is also proof of the existence of the hydrogen bond. All bond lengths and bond angles are in good agreement with the structure of the title compound. The dipole moment of the title compound is 2.57 Debye which indicates its polar nature.
Figure 6: Optimized molecular structure of the title compound at DFT/B3LYP method with a 6-311G (d,p) basis set
Table 1: Optimized geometrical parameters of the title compound at DFT/B3LYP method with a 6-311G(d,p) basis set
|
Bond lengths (Å)
|
|
C1-C2
|
1.3944
|
C6-H28
|
1.0821
|
C17-H20
|
1.0817
|
|
C1-C6
|
1.3901
|
C10-H11
|
1.0869
|
C18-C21
|
1.4038
|
|
C1-Cl29
|
1.7549
|
C10-C12
|
1.3453
|
C18-O26
|
1.3353
|
|
C2-C3
|
1.3862
|
C12-H13
|
1.0806
|
C19-C22
|
1.4019
|
|
C2-H7
|
1.0823
|
C12-C14
|
1.4784
|
C19-C23
|
1.083
|
|
C3-C4
|
1.4065
|
C14-O15
|
1.2441
|
C21-C22
|
1.3818
|
|
C3-H8
|
1.0832
|
C14-C16
|
1.4754
|
C21-H24
|
1.083
|
|
C4-C5
|
1.4047
|
C15-C27
|
1.6372
|
C22-H25
|
1.0845
|
|
C4-C10
|
1.4593
|
C16-C17
|
1.4109
|
C26-H27
|
0.9915
|
|
C5-C6
|
1.3896
|
C16-C18
|
1.4259
|
–
|
–
|
|
C5-H9
|
1.0844
|
C17-C19
|
1.3816
|
–
|
–
|
|
Bond angles (°)
|
|
C2-C1-C6
|
121.0327
|
C1-C6-H28
|
120.1921
|
C19-C17-H20
|
118.4684
|
|
C2-C1- Cl29
|
119.3779
|
C5-C6-H28
|
120.8569
|
C16-C18-C21
|
119.7607
|
|
C6-C1-Cl29
|
119.5894
|
C4-C10-H11
|
115.9548
|
C16-C18-C26
|
122.5821
|
|
C1-C2-C3
|
119.3428
|
C4-C10-C12
|
127.9173
|
C21-C18-O26
|
117.6572
|
|
C1-C2-H7
|
119.9106
|
H11-C10-C12
|
116.128
|
C17-C19-C22
|
119.3288
|
|
C3-C2-H7
|
120.7466
|
C10-C12-H13
|
120.9588
|
C17-C19-H23
|
120.2778
|
|
C2-C3-C4
|
121.2263
|
C10-C12-C14
|
120.091
|
C22-C19-H23
|
120.3934
|
|
C2-C3-H8
|
118.5822
|
H13-C12-C14
|
118.9503
|
C18-C21-C22
|
120.4437
|
|
C4-C3-H8
|
120.1915
|
C12-C14-O15
|
119.2758
|
C18-C21-H24
|
117.7904
|
|
C3-C4-C5
|
117.8397
|
C12-C14-C16
|
120.5044
|
C22-C21-H24
|
121.7658
|
|
C3-C4-C10
|
123.5496
|
O15-C14-C16
|
120.2199
|
C19-C22-C21
|
120.7024
|
|
C5-C4-C10
|
118.6107
|
C14-C16-C17
|
123.26
|
C19-C22-H25
|
119.8481
|
|
C4-C5-C6
|
121.6076
|
C14-C16-C18
|
118.7881
|
C21-C22-C25
|
119.4495
|
|
C4-C5-H9
|
119.1691
|
C17-C16-C18
|
117.9519
|
C18-O26-H27
|
106.3184
|
|
C6-C5-H9
|
119.2233
|
C16-C17-C19
|
121.8125
|
–
|
–
|
|
C1-C6-C5
|
118.951
|
C16-C17-H20
|
119.7192
|
–
|
–
|
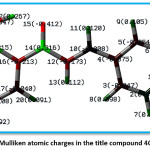
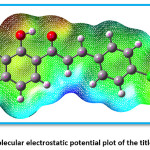
Figure 7 indicates the Mulliken charges in the title compound 4 CPHPP. The Mulliken charges are also presented in Table 2. From the Mulliken charges, it can be said that all the hydrogen atoms in the title compound are having a positive charge. This is also confirmed by the molecular electrostatic potential plot. The hydrogen atom which is the most electropositive in nature is H27 as it is attached to the electronegative oxygen atom. Then H7 is the second most electropositive due to the vicinity of the chlorine atom. Amongst all carbon atoms, C14 is the most electropositive in nature. This data is very important to decide the chemical reactivity of the title compound. The molecular electrostatic potential plot (MEP) is depicted in Figure 8. The MEP plot provides information regarding the chemical reactivity sites. The different colours in the MEP plots indicate zones of the positive, negative and neutral potentials. The MEP plot of the title compound indicates that the benzene ring bearing OH group would react more powerfully than the benzene ring bearing chlorine atom. Therefore, the electrophilic aromatic substitution reaction in the title compound is more favourable and selective in the former benzene ring. As predicted by the Mulliken atomic charges and MEP plot, the hydrogen atoms are present in the zones of positive electrostatic potential.
Figure 7: Mulliken atomic charges in the title compound 4CPHPP
Table 2: Mulliken atomic charges
|
Atom
|
Charge
|
Atom
|
Charge
|
Atom
|
Charge
|
|
1 C
|
-0.232848
|
11 H
|
0.120470
|
21 C
|
-0.088319
|
|
2 C
|
0.023941
|
12 C
|
-0.173175
|
22 C
|
-0.065451
|
|
3 C
|
-0.047129
|
13 H
|
0.112102
|
23 H
|
0.092261
|
|
4 C
|
-0.075108
|
14 C
|
0.315522
|
24 H
|
0.108219
|
|
5 C
|
-0.054460
|
15 O
|
-0.412054
|
25 H
|
0.101488
|
|
6 C
|
0.024372
|
16 C
|
-0.212886
|
26 O
|
-0.346990
|
|
7 H
|
0.122250
|
17 C
|
-0.039604
|
27 H
|
0.267205
|
|
8 H
|
0.097682
|
18 C
|
0.214894
|
28 H
|
0.124204
|
|
9 H
|
0.104927
|
19 C
|
-0.108467
|
29 Cl
|
-0.055737
|
|
10 C
|
-0.007885
|
20 H
|
0.090576
|
–
|
–
|
Figure 8: Molecular electrostatic potential plot of the title compound
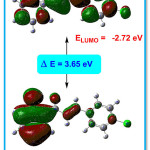
The HOMO-LUMO presentation of the title compound 4CPHPP is given in Figure 9. The imperative exploration on the electronic properties, such as HOMO and LUMO energies, was studied by the time-dependent DFT (TD-DFT) method at B3LYP/6-311G(d,p) basis set in the gas phase. The HOMO-LUMO energy gap in the title compound is 3.65 eV. The bandgap in the title compound suggests the inevitable charge transfer phenomenon is taking place within the title compound. From the HOMO-LUMO energies, various chemical reactivity parameters have been derived. The chemical potential (μ) and the charge transferred (∆Nmax) values are -4.545 eV and 2.49 eV respectively. The first singlet excitation energy in the title compound is 3.1579 eV and the absorption wavelength 392.62 nm with oscillator strength value (f) = 0.1798. The high value of absorption wavelength is due to presence of the extended conjugation which is known as the bathochromic shift. This excitation energy corresponds to the amount of energy required for the promotion of an electron from HOMO to LUMO.
Figure 9: HOMO-LUMO of the title compound 4CPHPP
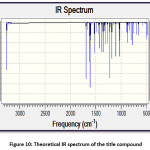
The theoretical and experimental IR spectra are presented in the Figure 10 and 3 respectively. The vibrational assignments were made by comparing the experimental FT-IR frequencies with the theoretical frequencies obtained by using B3LYP/6-311G(d,p) basis set. The very strong IR absorption peak at 1627.92 cm-1 is matching with the theoretical value 1625.28 cm-1 is assigned to C=O and C=C stretching vibrations. Another important experimental IR absorption peak is at 3441.01 cm-1 which is assigned to OH stretching vibrations is ideally matches the theoretical value which is 3450.35 cm-1. The decrease in both IR absorption frequencies is the consequence of the strong conjugation of the enone system with the aromatic rings. The theoretical IR signal at 1535.83 1504.18 cm-1 is due to the Ar C=C stretching vibrations and it is correlated with the experimental value 1504.18 cm-1. Other vibrational data is also in good agreement with the experimental FT-IR frequencies. Thermochemical data obtained from the harmonic frequencies are tabulated in Table 3. The important thermochemical parameters like total Energy (thermal), total heat capacity (Cv), and entropy (S) are evaluated. The entropy value indicates the degree of freedom whereas the total energy suggests the thermal stability of the title compound. The thermodynamic data could be used to predict the other thermochemical parameters also.
Figure 10: Theoretical IR spectrum of the title compound
Table 3: Thermochemical data of the title compound
|
Parameters
|
Translational
|
Rotational
|
Vibrational
|
Total
|
|
Total Energy (Thermal) KCal/Mol
|
0.889
|
0.889
|
145.279
|
147.056
|
|
Total heat capacity (Cv) Cal/Mol/Kelvin
|
2.981
|
2.981
|
52.009
|
57.971
|
|
Entropy (S) Cal/Mol/Kelvin
|
42.544
|
34.242
|
51.6031
|
128.389
|
Conclusion
In conclusion, (E)-3-(4-chlorophenyl)-1-(2-hydroxyphenyl)prop-2-en-1-one has been explored to study its structural, electronic, and chemical viewpoints. The conclusion of the present research is summarized as-
- The (E)-3-(4-chlorophenyl)-1-(2-hydroxyphenyl)prop-2-en-1-one has been synthesized and characterized by FT-IR, 1HNMR, and 13C NMR spectral techniques. DFT method at B3LYP/6-311G(d,p) basis set has been employed.
- The detailed investigation of bond lengths and bond angles is discussed to comprehend the geometrical framework.
- To explore its chemical behaviour, Mulliken atomic charges, molecular electrostatic potential surface, and electronic parameters are introduced.
- The imperative exploration on the electronic properties, such as HOMO and LUMO energies, was studied by the time-dependent DFT (TD-DFT) method.
- The dipole moment of the title molecule is 2.57 Debye with C1 point group symmetry.
- The most electropositive carbon and hydrogen atoms in the title molecule are C14 and H27 respectively. Amongst aromatic C=C, the C16-C18 is the longest, and C17-C19 is the shortest bond.
- The molecular electrostatic potential surface analysis suggested the positive electrostatic potential is around hydrogen atoms.
- The vibrational assignments in the title compound were done by comparing the experimental IR frequencies with the scaled frequencies obtained using computational work. Additionally, some noteworthy thermochemical data is acquired utilizing a similar basis set using frequencies.
Acknowledgments
Authors acknowledge Sophisticated Analytical Instrument Facility, Punjab University, Chandigarh for NMR and CIC, KTHM College, Nashik for FT-IR spectral analysis. The authors also would like to thank Principals of Arts, Science and Commerce College, Nandgaon and Arts, Science and Commerce College, Manmad, for permission and providing necessary research facilities. The authors are very grateful to Prof. Arun B. Sawant for his generous help in the Gaussian guidance. Dr. Aapoorva Prashant Hiray, Coordinator, MG Vidyamandir Institute, is gratefully acknowledged for Gaussian package.
Funding
No funding was received to carry out the research work presented in this research paper.
Conflict of Interest
The authors declare that they do not have any conflict of interest.
References
- M. J. Matos, S. Vazquez-Rodriguez, E. Uriarte, L. Santana, Potential pharmacological uses of chalcones: a patent review (from June 2011–2014), Expert Opin. Ther. Pat. 25(3), 351-366 (2015).
- A. Modzelewska, C. Pettit, G. Achanta, N. E. Davidson, P. Huang, S. R. Khan. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg. Med. Chem.. 14(10), 3491-3495 (2006).
- C. Jin, Y. J. Liang, H. He, L. Fu, Synthesis and antitumor activity of novel chalcone derivatives. Biomed. Pharmacother. 67(3), 215-217 (2013).
- D. I. Ugwu, B. E. Ezema, U. C. Okoro, F. U. Eze, O. C. Ekoh, M. C. Egbujor, D. I. Ugwuja, Syntheses and pharmacological applications of chalcones: a review. Int. J. Chem. Sci. 13(1), 459-500 (2015).
- S. U. Rizvi, H. L. Siddiqui, M. Johns, M. Detorio, R. F. Schinazi, Anti-HIV-1 and cytotoxicity studies of piperidyl-thienyl chalcones and their 2-pyrazoline derivatives. Med. Chem. Res. 21(11), 3741-3749 (2012).
- G. Mazzone, N. Malaj, A. Galano, N. Russo, M. Toscano, Antioxidant properties of several coumarin–chalcone hybrids from theoretical insights. RSC Adv. 5(1), 565-575 (2015).
- K. V. Sashidhara, S. R. Avula, V. Mishra, G. R. Palnati, L. R. Singh, N. Singh, Y. S. Chhonker, P. Swami, R. S. Bhatta, Identification of quinoline-chalcone hybrids as potential antiulcer agents. Eur. J. Med. Chem. 89, 638-653 (2015).
- R. Raj, A. Saini, J. Gut, P. J. Rosenthal, V. Kumar, Synthesis and in vitro antiplasmodial evaluation of 7-chloroquinoline–chalcone and 7-chloroquinoline–ferrocenylchalcone conjugates. Eur. J. Med. Chem. 95, 230-239 (2015).
- D. K. Mahapatra, S. K. Bharti, Therapeutic potential of chalcones as cardiovascular agents. Life Sci. 148, 154-172 (2016).
- M. Mujahid, P. Yogeeswari, D. Sriram, U. M. Basavanag, E. Díaz-Cervantes, L. Córdoba-Bahena, J. Robles, R. G. Gonnade, M. Karthikeyan, R. Vyas, M. Muthukrishnan , Spirochromone-chalcone conjugates as antitubercular agents: synthesis, bio evaluation and molecular modeling studies. RSC Adv. 5(129), 106448-10660 (2015).
- W. C. Chu, P. Y. Bai, Z. Q. Yang, D. Y. Cui, Y. G. Hua, Y. Yang, Q. Q. Yang, E. Zhang, S. Qin, Synthesis and antibacterial evaluation of novel cationic chalcone derivatives possessing broad spectrum antibacterial activity. Eur. J. Med. Chem. 143, 905-921 (2018).
- Y. Zheng, X. Wang, S. Gao, M. Ma, G. Ren, H. Liu, X. Chen, Synthesis and antifungal activity of chalcone derivatives. Nat. Prod. Res. 29(19), 1804-1810 (2015).
- D. Zhou, D. Xie, F. He, B. Song, D. Hu, Antiviral properties and interaction of novel chalcone derivatives containing a purine and benzenesulfonamide moiety. Bioorg. Med. Chem. Lett. 28(11), 2091-2097 (2018).
- D. K. Mahapatra, S. K. Bharti, V. Asati, Chalcone derivatives: Anti-inflammatory potential and molecular targets perspectives. Curr. Top. Med. Chem. 17(28), 3146-3169 (2017).
- C. S. Sharma, K. S. Shekhawat, C. S. Chauhan, N. Kumar, Synthesis and anticonvulsant activity of some chalcone derivatives. J. Chem. Pharm. Res. 5(10), 450-454 (2013).
- V. A. Adole, T. B. Pawar, B. S. Jagdale, Aqua-mediated rapid and benign synthesis of 1,2,6,7-tetrahydro-8H-indeno[5,4-b]furan-8-one-appended novel 2-arylidene indanones of pharmacological interest at ambient temperature J. Chin. Chem. Soc. 67, 306-315 (2020).
- R. Kalirajan, S. U. Sivakumar, S. Jubie, B. Gowramma, B. Suresh, Synthesis and biological evaluation of some heterocyclic derivatives of chalcones. Int. J. ChemTech Res. 1(1), 27-34 (2009).
- M. J. Elarfi, H. A. Al-Difa, Synthesis of some heterocyclic compounds derived from chalcones. Sci. Rev. Chem. Commun. 2(2), 103-107 (2012).
- M. V. Jyothi, Y. R. Prasad, P. Venkatesh, M. Sureshreddy, Synthesis and antimicrobial activity of some novel chalcones of 3-acetyl pyridine and their pyrimidine derivatives. Chem. Sci. Trans. 1(3), 716-722 (2012).
- S. Fandakli, N. Kahriman, T. B. YÜCEL, Ş. A. KARAOĞLU, N. Yayli, Biological evaluation and synthesis of new pyrimidine-2 (1H)-ol/-thiol derivatives derived from chalcones using the solid phase microwave method. Turk. J. Chem. 42(2), 520-535 (2018).
- V. D. Joshi, M. D. Kshirsagar, S. Sarita, Synthesis and antimicrobial activities of various pyrazolines from chalcones. Int. J. ChemTech Res. 4(3), 971-975 (2012).
- A. H. Adel, E. S. Ahmed, M. A. Hawata, E. R. Kasem, M. T. Shabaan, Synthesis and antimicrobial evaluation of some chalcones and their derived pyrazoles, pyrazolines, isoxazolines, and 5,6-dihydropyrimidine-2-(1H)-thiones. Monatsh. Chem. 138(9), 889-897 (2007).
- F. L. Ansari, S. Umbreen, L. Hussain, T. Makhmoor, S. A. Nawaz, M. A. Lodhi, S. N. Khan, F. Shaheen, M. I. Choudhary, Syntheses and Biological Activities of Chalcone and 1,5‐Benzothiazepine Derivatives: Promising New Free‐Radical Scavengers, and Esterase, Urease, and α‐Glucosidase Inhibitors. Chem. Biodiversity. 2(4), 487-496 (2005).
- A. Atac, M. Karabacak, C. Karaca, E. Kose, NMR, UV, FT-IR, FT-Raman spectra and molecular structure (monomeric and dimeric structures) investigation of nicotinic acid N-oxide: A combined experimental and theoretical study. Spectrochim. Acta, Part A. 85(1), 145-154 (2012).
- F Chain, E. Romano, P. Leyton, C. Paipa, C. A. Catalán, M. A. Fortuna, S. A. Brandán, An experimental study of the structural and vibrational properties of sesquiterpene lactone cnicin using FT-IR, FT-Raman, UV–visible and NMR spectroscopies. J. Mol. Struct. 1065, 160-169 (2014).
- E. Kose, A. Atac, M. Karabacak, P. B. Nagabalasubramanian, A. M. Asiri, S. Periandy, FT-IR and FT-Raman, NMR and UV spectroscopic investigation and hybrid computational (HF and DFT) analysis on the molecular structure of mesitylene. Spectrochim. Acta, Part A. 116, 622-634 (2013).
- J. B. Bhagyasree, H. T. Varghese, C. Y. Panicker, J. Samuel, C. Van Alsenoy, K Bolelli, I. Yildiz, E. Aki, Vibrational spectroscopic (FT-IR, FT-Raman, 1H NMR and UV) investigations and computational study of 5-nitro-2-(4-nitrobenzyl) benzoxazole. Spectrochim. Acta, Part A. 102, 99-113 (2013).
- V. A. Adole, B. S. Jagdale, T. B. Pawar, A. B. Sawant, Experimental and theoretical exploration on single crystal, structural, and quantum chemical parameters of (E)‐7‐(arylidene)‐1,2,6,7‐tetrahydro‐8 H‐indeno [5,4‐b] furan‐8‐one derivatives: A comparative study. Journal of the Chinese Chemical Society. https://doi.org/10.1002/jccs.202000006
- M. Karabacak, E. Kose, A Atac, A. M. Asiri, M. Kurt, Monomeric and dimeric structures analysis and spectroscopic characterization of 3,5-difluorophenylboronic acid with experimental (FT-IR, FT-Raman, 1H and 13C NMR, UV) techniques and quantum chemical calculations. J. Mol. Struct. 1058, 79-96 (2014).
- S. Ramalingam, M. Karabacak, S. Periandy, N. Puviarasan, D. Tanuja, Spectroscopic (infrared, Raman, UV and NMR) analysis, Gaussian hybrid computational investigation (MEP maps/HOMO and LUMO) on cyclohexanone oxime. Spectrochim. Acta, Part A. 96, 207-220 (2012).
- D. Shoba, S. Periandi, S. Boomadevi, S Ramalingam, E. Fereyduni, FT-IR, FT-Raman, UV, NMR spectra, molecular structure, ESP, NBO and HOMO–LUMO investigation of 2-methylpyridine 1-oxide: A combined experimental and DFT study. Spectrochim. Acta, Part A. 118, 438-447 (2014).
- A. Suvitha, S. Periandy, S. Boomadevi, M. Govindarajan, Vibrational frequency analysis, FT-IR, FT-Raman, ab initio, HF and DFT studies, NBO, HOMO–LUMO and electronic structure calculations on pycolinaldehyde oxime. Spectrochim. Acta, Part A. 117, 216-224 (2014).
- M. Kalinowska, R. Świsłocka, W. Lewandowski, The spectroscopic (FT-IR, FT-Raman, UV and 1H, 13C NMR) and theoretical studies of alkali metal o-methoxybenzoates. J. Mol. Struct. 792, 130-138 (2006).
- M. Raja, R. R. Muhamed, S. Muthu, M. Suresh, Synthesis, spectroscopic (FT-IR, FT-Raman, NMR, UV–Visible), first order hyperpolarizability, NBO and molecular docking study of (E)-1-(4-bromobenzylidene) semicarbazide. J. Mol. Struct. 1128, 481-492 (2017).
- S Sudha, N. Sundaraganesan, K Vanchinathan, K. Muthu, S. P. Meenakshisundaram, Spectroscopic (FTIR, FT-Raman, NMR and UV) and molecular structure investigations of 1,5-diphenylpenta-1,4-dien-3-one: A combined experimental and theoretical study. J. Mol. Struct. 1030, 191-203 (2012).
- R. Kumar, A. Kumar, V. Deval, A. Gupta, P. Tandon, P. S. Patil, P. Deshmukh, D. Chaturvedi, J. G. Watve, Molecular structure, spectroscopic (FT-IR, FT Raman, UV, NMR and THz) investigation and hyperpolarizability studies of 3-(2-Chloro-6-fluorophenyl)-1-(2-thienyl) prop-2-en-1-one. J. Mol. Struct. 1129, 292-304 (2017).
- V. A. Adole, R. H. Waghchaure, B. S. Jagdale, T. B. Pawar, Investigation of Structural and Spectroscopic Parameters of Ethyl 4-(4-isopropylphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate: a DFT Study. Chemistry & Biology Interface, 10: (2020)
- A. B. Sawant, R. S. Nirwan, Synthesis, characterization and DFT studies of 6, 8-dichloro-2-(4-chlorophenyl)-4H-chromen-4-one, Indian J. Pure Appl. Phys. 50(5), 308-313 (2012).
- A. B. Sawant, C. H. Gill, R. S. Nirwan, Molecular structure and vibrational spectra of 2-[5-(4-chlorophenyl)-4, 5-dihydro-1H-pyrazol-3-yl] phenol, Indian J. Pure Appl. Phys. 50(1):38-44 (2012).
- P.B.Koli, K.H.Kapadnis, and U.G.Deshpande, Transition metal decorated Ferrosoferric oxide (Fe3O4): An expeditious catalyst for photodegradation of Carbol Fuchsin in environmental remediation. Journal of Environmental Chemical Engineering, 7(5), p.103373 (2019)
- P.B.Koli, K.H Kapadnis, U.G. Deshpande and M.R. Patil, Fabrication and characterization of pure and modified Co3O4 nanocatalyst and their application for photocatalytic degradation of eosine blue dye: a comparative study. Journal of Nanostructure in Chemistry, 8(4), 453-463 (2018)
- P.B.Koli, K.H. Kapadnis and U.G. Deshpande, Nanocrystalline-modified nickel ferrite films: an effective sensor for industrial and environmental gas pollutant detection. Journal of Nanostructure in Chemistry, 9(2), 95-110 (2019)
- P.B.Koli, K.H. Kapadnis, U.G.Deshpande, B.P. More and U.J.Tupe, Sol-Gel Fabricated Transition Metal Cr3+, Co2+ Doped Lanthanum Ferric Oxide (LFO-LaFeO3) Thin Film Sensors for the Detection of Toxic, Flammable Gases: A Comparative Study. Material Science Research India, 17(1), 70-83 (2020)
- V. A. Adole, T. B. Pawar, P. B. Koli, B. S. Jagdale, Exploration of catalytic performance of nano-La2O3 as an efficient catalyst for dihydropyrimidinones/thione synthesis and gas sensing. J. Nanostructure Chem. 9:61-76 (2019)
- V. A. Adole, B. S. Jagdale, T. B. Pawar, A. A. Sagane, Ultrasound promoted stereoselective synthesis of 2, 3-dihydrobenzofuran appended chalcones at ambient temperature. S. Afr. J. Chem. 73(1), 35-43 (2020).
- M. J. Frisch, G. W. Trucks, H. B. Schlegal, et al., G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven,K. N. Kudin, J. C. Burant, J. M. Millam,Gaussian 03, revision E.01, Gaussian, Inc., Wellingford CT, (2004)
Views: 1,552
 This work is licensed under a Creative Commons Attribution 4.0 International License.
This work is licensed under a Creative Commons Attribution 4.0 International License.