Molecular Structure, Electronic, Chemical and Spectroscopic (UV-Visible and IR) Studies of 5-(4-Chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-pyrazole: Combined DFT and Experimental Exploration
Sandip S. Pathade1 , Vishnu A. Adole2, Bapu S. Jagdale2, Thansing B. Pawar3*
, Vishnu A. Adole2, Bapu S. Jagdale2, Thansing B. Pawar3*
1Department of Chemistry, Maharaja Sayajirao Gaikwad Arts, Science and Commerce College Malegaon, (Affiliated to SP Pune University) Nashik-423 105, India
2Department of Chemistry, Arts, Science and Commerce College, Manmad, (Affiliated to SP Pune University) Nashik-423 104, India
3Department of Chemistry, Loknete Vyankatrao Hiray Arts, Science and Commerce College Panchavati, (Affiliated to SP Pune University) Nashik-422 003, India
Corresponding Author Email: Pawartbpawar03@gmail.com
DOI : http://dx.doi.org/10.13005/msri.17.special-issue1.05
Article Publishing History
Article Received on : 21 June 2020
Article Accepted on : 25 July 2020
Article Published : 27 Jul 2020
Plagiarism Check: Yes
Reviewed by: Dr Fakruddin Shaik
Second Review by: Ayyappan Elangovan
Final Approval by: Arunabha Roy
Article Metrics
ABSTRACT:
The current examination deals with a detailed investigation on the computational study of 5-(4-chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-pyrazole (CPMPP) by using density functional theory (DFT). CPMPP is synthesized and characterized by UV-Visible, FT-IR, 1H NMR, and 13C NMR spectroscopic methods. The molecular structure, optimized geometrical parameters, and vibrational assignments have been established employing the DFT method, the B3LYP method, and the 6-311++G (d,p) basis set. Frontier molecular orbital (FMO) analysis and various global reactivity parameters are also discussed for the better comprehension of the chemical reactivity. Theoretical and Experimental; UV-Visible analysis is compared and a good deal of agreement is found. Experimental vibrational frequencies were compared with the theoretical IR spectrum to mark the correct vibrational assignments. Molecular electrostatic surface potential (MESP) and Mulliken atomic charges are computed at the same level of theory to locate the charge density. Absorption energies, excitation energy, oscillator strength, and transitions have been computed at TD-B3LYP/6-311++G (d,p) level of theory for B3LYP/6-311++G (d,p) optimized geometry.
KEYWORDS:
5-(4-Chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-pyrazole; DFT; Gaussian; Molecular Structure; Vibrational Assignments
Copy the following to cite this article:
Pathade S. S, Adole V. A, Jagdale B. S, Pawar T. B. Molecular Structure, Electronic, Chemical and Spectroscopic (UV-Visible and IR) Studies of 5-(4-Chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-pyrazole: Combined DFT and Experimental Exploration. Mat. Sci. Res. India; Special Issue (2020).
|
Copy the following to cite this URL:
Pathade S. S, Adole V. A, Jagdale B. S, Pawar T. B. Molecular Structure, Electronic, Chemical and Spectroscopic (UV-Visible and IR) Studies of 5-(4-Chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-pyrazole: Combined DFT and Experimental Exploration. Mat. Sci. Res. India; Special Issue (2020). Available from: https://bit.ly/2OVQ0aW
|
Introduction
Pyrazoles are five-membered heterocycles that establish a class of heterocyclic compounds especially valuable in organic synthesis. Pyrazoline is one of the imperative synthons in therapeutic science and has played a crucial role in the development of heterocyclic compounds. The chalcones are significant intermediates for the synthesis of various heterocyclic compounds including pyrazolines. Pyrazole frameworks have pulled in more consideration because of their fascinating pharmacological properties.1-3 The wide range of biological properties includes anticancer activity,4 anti-tubercular,5 antibacterial,6 antifungal,7 anti-inflammatory,8 analgesic,9 anti-viral activity,10 anti-diabetic,11 anti-malarial,12 etc. Few examples of biologically potent drugs containing pyrazole structure are given in Figure 1. Lonazolac is found to show excellent anti-inflammatory activity in disease treatments whereas fezolamine shows anti-depressant and difenamizole shows analgesic activity. The green chemistry has advanced in recent years and explored for the synthesis of variety of organic compounds having potential biological applications.13-20 The PEG solvents have been used to increase environmental sustainability and energy-efficient reactions.21-23
Theoretical chemistry based on DFT can predict various molecular properties. By virtue of DFT, different spectroscopic assessments can be achieved; UV-Visible spectra, IR and Raman frequencies, NMR chemical shifts, and spin-spin coupling constants.24-32 Also, DFT calculations can anticipate FMO energies, bond lengths, bond angles, dihedral angles, absorption energies, etc.33-45 The comparison of theoretical calculations with experimental outcomes gives decent information. Critically, the examination of the reaction mechanisms is made simpler because of calculation estimations in view of DFT. Considering all these critical angles and continuation of our enthusiasm for synthesizing heterocyclic compounds, we wish to report synthesis and density functional theory investigation of 5-(4-chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-pyrazole. In the current investigation various structural, spectroscopic, and quantum chemical facets of title compound have been revealed.
Experimental
Material and Methods
All the chemicals needed for synthesis were obtained from a commercial source (AR grade with purity > 99%) and used without further purification. Melting points of the compounds were determined in an open capillary tube and is uncorrected, IR spectra were recorded on the Shimadzu FT-IR spectrometer using potassium bromide pellets. 1H NMR was determined on Bruker Advance II 500 MHz spectrometer using DMSO as a solvent. The UV-Visible analysis was performed within 200 to 800 nm range in the DMSO solvent. The reactions were monitored by thin-layer chromatography (TLC, Merck) using aluminum sheets coated with silica gel by using n-hexane and ethyl acetate as an eluent.
Synthesis of Chalcone
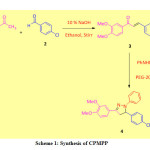
An equimolar mixture of 3,4-dimethoxy acetophenone (1, 0.01 mol) and 4-chloro benzaldehyde (2, 0.01 mol), and 10 % NaOH (10 mL) in ethanol (15 mL) was stirred together for 14 hours at room temperature. The completion of the reaction was monitored by TLC. Then the reaction mixture was poured into crushed ice and acidified with dilute HCl. The solid obtained was filtered, washed with water, dried, and recrystallized from ethanol to obtain desire chalcone.
Synthesis of CPMPP
The solution of chalcone 3 (20 mmol) and phenylhydrazine (20 mmol), and 10% NaOH in PEG-200 (10 mL) were refluxed for 2 hours at 70-80 ℃. The completion of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was poured into crushed ice and precipitate obtained was filtered under vacuum, washed with water, dried, and recrystallized from ethanol to furnish the title compound 4. The overall reaction is given in Scheme 1.
Physicochemical and Spectral Analysis
Light yellowish solid; Yield: 92 %; m.p. 140-142 ℃. FT-IR (KBr, in cm-1): 1596 (C=N str), 2917 (Ar-H str), 1495 (Aro. C=C str), 816 (Ar-Cl str); 1H NMR (500 MHz, CDCl3) δ: 7.49 (d, J = 2.0 Hz, 1H), 7.30 (d, J = 8.5 Hz, 2H), 7.26 (d, J = 6.5 Hz, 2H), 7.20 – 7.16 (m, 2H), 7.02 (m, 3H), 6.84 (d, J = 8.4 Hz, 1H), 6.82 – 6.75 (m, 1H), 5.21 (dd, J = 12.3, 7.2 Hz, 1H), 3.98 (s, 3H), 3.90 (s, 3H), 3.82 (dd, J = 16.9, 12.3 Hz, 1H), 3.08 (dd, J = 16.9, 7.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ (ppm): 150.01, 149.17, 146.90, 144.88, 141.20, 133.30, 129.35, 128.98, 127.36, 125.57, 119.19, 119.11, 113.32, 110.62, 108.09, 63.93, 55.97, 43.68.
Computational Details
All the computational calculations are determined in the gas phase by the DFT method using Gaussian-03(W) software. To analyze the theoretical parameters of the titled compound, geometry was optimized by DFT/B3LYP method with a 6-311++G (d,p) basis set [46]. The optimized structure of compounds was used for the calculation of thermodynamic parameters, global chemical reactivity parameters, frontier molecular orbital analysis, and plotting the molecular electrostatic potentials. Absorption energies, excitation energy, oscillator strength, and transitions have been computed at TD-B3LYP/6-311++G (d,p) level of theory for B3LYP/6-311++G (d,p) optimized geometry.
Results and Discussion
Structural Analysis
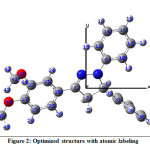
The optimized molecular structure of the CPMPP is given in Figure 2. Optimized bond lengths and bond angles of CPMPP at B3LYP/6-311++G (d,p) are presented in Table 1. The CPMPP possesses aromatic C=C bond lengths from 1.38 Å to 1.40 Å. The azo group (N7-N8) bond is 1.3708 Å long and the imine (C1=N8) bond is 1.2887 Å in length. The dipole moment of the CPMPP is 4.59 Debye with C1 point group symmetry and -1609.51 a.u. E(B3LYP) energy. The bond angles N8-C1-C20, C1-N8-N7, O40-C45-H47, and C32-C36-Cl49 are 122.6027°, 110.3769°, 111.2483°, and 119.4557° respectively. Other bond length and bond angle data are also in good agreement.
Table 1: Optimized bond lengths and bond angles at B3LYP/6-311++G (d,p) level
|
Bond length (Å)
|
|
C1-C2
|
1.5164
|
C12-C16
|
1.3919
|
C29-C31
|
1.3970
|
|
C1-N8
|
1.2887
|
C12-H17
|
1.0846
|
C30-C32
|
1.3933
|
|
C1-C20
|
1.4612
|
C14-C16
|
1.3960
|
C30-H33
|
1.0857
|
|
C2-C3
|
1.5542
|
C14-H18
|
1.0847
|
C31-C34
|
1.3923
|
|
C2-H4
|
1.0930
|
C16-H19
|
1.0835
|
C31-H35
|
1.0840
|
|
C2-H5
|
1.0931
|
C20-C21
|
1.4084
|
C32-C36
|
1.3904
|
|
C3-H6
|
1.0962
|
C20-C22
|
1.3977
|
C32-H37
|
1.0825
|
|
C3-N7
|
1.4793
|
C21-C23
|
1.3820
|
C34-C36
|
1.3915
|
|
C3-C29
|
1.5202
|
C21-H24
|
1.0824
|
C34-H38
|
1.0826
|
|
N7-N8
|
1.3708
|
C22-C25
|
1.3960
|
C36-Cl49
|
1.7595
|
|
N7-C9
|
1.3987
|
C22-H26
|
1.0835
|
O39-C41
|
1.4325
|
|
C9-C10
|
1.4051
|
C23-C27
|
1.4165
|
O40-C45
|
1.4223
|
|
C9-C11
|
1.4070
|
C23-O39
|
1.3718
|
C41-H42
|
1.0895
|
|
C10-C12
|
1.3932
|
C25-C27
|
1.3936
|
C41-H43
|
1.0920
|
|
C10-H13
|
1.0815
|
C25-H28
|
1.0817
|
C41-H44
|
1.0957
|
|
C11-C14
|
1.3889
|
C27-O40
|
1.3616
|
C45-H46
|
1.0886
|
|
C11-H15
|
1.0805
|
C29-C30
|
1.3969
|
C45-H47
|
1.0950
|
|
–
|
–
|
–
|
–
|
C45-H48
|
1.0953
|
|
Bond angle (°)
|
|
C2-C1-N8
|
112.7333
|
C10-C12-H17
|
118.9366
|
C29-C30-C32
|
121.1396
|
|
C2-C1-C20
|
124.662
|
C16-C12-H17
|
120.0987
|
C29-C30-H33
|
119.8456
|
|
N8-C1-C20
|
122.6027
|
C11-C14-C16
|
121.1398
|
C32-C30-H33
|
119.0141
|
|
C1-C2-C3
|
102.4226
|
C11-C14-H18
|
118.9236
|
C29-C31-C34
|
120.8973
|
|
C1-C2-H4
|
111.7172
|
C16-C14-H18
|
119.9358
|
C29-C31-H35
|
119.5406
|
|
C1-C2-H5
|
111.6775
|
C12-C16-C14
|
118.7769
|
C34-C31-H35
|
119.5533
|
|
C3-C2-H4
|
112.2421
|
C12-C16-H19
|
120.5992
|
C30-C32-C36
|
118.9867
|
|
C3-C2-H5
|
111.3691
|
C14-C16-H19
|
120.6239
|
C30-C32-H37
|
120.8066
|
|
H4-C2-H5
|
107.4636
|
C1-C20-C21
|
120.9602
|
C36-C32-H37
|
120.2055
|
|
C2-C3-H6
|
110.4550
|
C1-C20-C22
|
121.0493
|
C31-C34-C36
|
119.2429
|
|
C2-C3-N7
|
101.8185
|
C21-C20-C22
|
117.9901
|
C31-C34-H38
|
120.6664
|
|
C2-C3-C29
|
112.9459
|
C20-C21-C23
|
121.5549
|
C36-C34-H38
|
120.0897
|
|
H6-C3-N7
|
109.1755
|
C20-C21-H24
|
120.0735
|
C32-C36-C34
|
121.0291
|
|
H6-C3-C29
|
108.6815
|
C23-C21-H24
|
118.3982
|
C32-C36-Cl49
|
119.4557
|
|
N7-C3-C29
|
113.5987
|
C20-C22-C25
|
121.0966
|
C34-C36-Cl49
|
119.5142
|
|
C3-N7-N8
|
112.5815
|
C20-C22-H26
|
120.4259
|
C23-O39-C41
|
116.2329
|
|
C3-N7-C9
|
124.3649
|
C25-C22-H26
|
118.4773
|
C27-O40-C45
|
118.5520
|
|
N8-N7-C9
|
119.4649
|
C21-C23-C27
|
119.8929
|
O39-C41-H42
|
105.9929
|
|
C1-N8-N7
|
110.3769
|
C21-C23-O39
|
118.8338
|
O39-C41-H43
|
111.3279
|
|
N7-C9-C10
|
120.5477
|
C27-C23-O39
|
121.1526
|
O39-C41-H44
|
110.4094
|
|
N7-C9-C11
|
120.6239
|
C22-C25-C27
|
120.5002
|
H42-C41-H43
|
109.6557
|
|
C10-C9-C11
|
118.8234
|
C22-C25-H28
|
119.0783
|
H42-C41-H44
|
109.3871
|
|
C9-C10-C12
|
120.2049
|
C27-C25-H28
|
120.4191
|
H43-C41-H44
|
109.9796
|
|
C9-C10-H13
|
120.6123
|
C23-C27-C25
|
118.9595
|
O40-C45-H46
|
105.7824
|
|
C12-C10-H13
|
119.1746
|
C23-C27-O40
|
116.2956
|
O40-C45-H47
|
111.2483
|
|
C9-C11-C14
|
120.0902
|
C25-C27-O40
|
124.7425
|
O40-C45-H48
|
111.4068
|
|
C9-C11-H15
|
119.1303
|
C3-C29-C30
|
119.9300
|
H46-C45-H47
|
109.3764
|
|
C14-C11-H15
|
120.7787
|
C3-C29-C31
|
121.3116
|
H46-C45-H48
|
109.3962
|
|
C10-C12-C16
|
120.9644
|
C30-C29-C31
|
118.7030
|
H47-C45-H48
|
109.5428
|
Charge Density Distribution Study
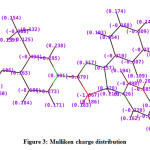
Mulliken atomic charges and molecular electrostatic surface potential are studied for the investigation of a charge density distribution study. Mulliken atomic charges are presented in Table 2 and Figure 3. The C2 carbon atom is the most electronegative with a charge density of -1.06730 whereas C20 carbon atom is the most electropositive carbon atom with charge density of 0.90122. Talking about hydrogen atoms, all hydrogen atoms are electropositive.
Table 2: Mulliken atomic charges
|
Atom
|
Charge
|
Atom
|
Charge
|
Atom
|
Charge
|
|
C1
|
-0.47944
|
H18
|
0.17408
|
H35
|
0.19404
|
|
C2
|
-1.06730
|
H19
|
0.14055
|
C36
|
0.25209
|
|
C3
|
0.22775
|
C20
|
0.90122
|
H37
|
0.20442
|
|
H4
|
0.18280
|
C21
|
-0.29498
|
H38
|
0.20834
|
|
H5
|
0.18648
|
C22
|
-0.27261
|
O39
|
-0.12454
|
|
H6
|
0.21757
|
C23
|
-0.49427
|
O40
|
-0.18626
|
|
N7
|
0.15700
|
H24
|
0.23809
|
C41
|
-0.30824
|
|
N8
|
0.51666
|
C25
|
-0.18568
|
H42
|
0.15422
|
|
C9
|
-0.31043
|
H26
|
0.17104
|
H43
|
0.15928
|
|
C10
|
-0.30652
|
C27
|
0.16294
|
H44
|
0.13171
|
|
C11
|
0.05379
|
H28
|
0.18358
|
C45
|
-0.31298
|
|
C12
|
-0.23681
|
C29
|
0.42632
|
H46
|
0.18028
|
|
H13
|
0.10948
|
C30
|
-0.22009
|
H47
|
0.15328
|
|
C14
|
-0.16792
|
C31
|
-0.49845
|
H48
|
0.16010
|
|
H15
|
0.19277
|
C32
|
-0.38618
|
Cl49
|
0.46538
|
|
C16
|
-0.30929
|
H33
|
0.16241
|
–
|
–
|
|
H17
|
0.17885
|
C34
|
-0.68456
|
–
|
–
|
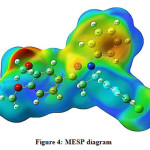
The MESP plot is depicted in Figure 4. The molecular electrostatic potential surface analysis could provide information about the crucial molecular properties like dipole moment, partial charges, and chemical reactivity of the molecules. The presence of various distinct colors indicates the variation in the electron density distribution. The red and yellow colors speak about high electron density, blue and green colors indicate regions with positive and zero electrostatic potentials respectively. The present study of the MESP plot indicates that the phenyl ring attached to a nitrogen atom is highly susceptible to react with the electrophilic reagents and therefore the molecule could undergo electrophilic aromatic substitution reactions at the phenyl ring attached to a nitrogen atom.
FMO, Electronic and Chemical Reactivity Studies
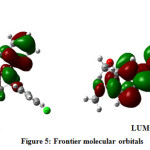
The pictorial representation of the HOMO-LUMO of the title compound is given in Figure 5. The data of the electronic parameters and the global reactivity descriptors are given in Table 3. The HOMO-LUMO orbitals are called as Frontier Molecular Orbitals (FMO). The energy gap between HOMO-LUMO is a fundamental pointer of kinetic stability. The assessment of the wave function indicates that the electron absorption corresponds to the transition from the HOMO to the LUMO and is mostly portrayed by one electron promotion. The Koopmans’ hypothesis has been utilized for the examination of the global reactivity descriptor parameters and because of frontier molecular orbital energies, the different parameters such as ionization potential (I), electron affinity (A), band gap (Eg), electronegativity (χ), absolute hardness (ɳ), global softness (S), global electrophilicity (ω), chemical potential (µ), and maximum no. of electron transferred (ΔNmax) have been built up.
Table 3: Global chemical reactivity descriptors
|
Molecular Properties
|
Value
|
|
EHOMO
|
-5.1838 eV
|
|
ELUMO
|
-1.3954 eV
|
|
∆E (ELUMO-EHOMO)
|
3.7884 eV
|
|
Ionisation energy (I)
|
5.1838 eV
|
|
Electron affinity (EA)
|
1.3954 eV
|
|
Electronegativity (χ)
|
3.2896 eV
|
|
Chemical hardness (η)
|
1.8942 eV
|
|
Chemical softness (S)
|
0.5279 eV-1
|
|
Chemical potential (µ)
|
-3.2896 eV
|
|
Global electrophilicity index (ω)
|
2.8565 eV
|
|
Maximum number of electrons transferred (∆Nmax)
|
1.7367 eV
|
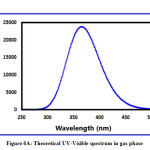
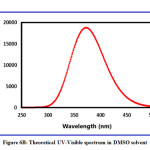
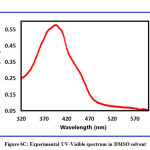
The exploration suggests that the title compound has a low band energy gap and therefore will ease in the charge transfer phenomenon within the molecule. The maximum charge transfer is occurring in the molecule is 1.7367 eV. The global electrophilicity index is = 2.8565 eV. Absorption energies (λ in nm), excitation energy (eV), oscillator strength (ƒ) and transitions have been established by computing the results at TD-B3LYP/6-311++G (d,p) level of theory for B3LYP/6-311++G (d,p) optimized geometries and this data is furnished in Table 4. The theoretical UV-Visible spectrum is given in Figure 6A (gas phase) and Figure 6B (DMSO). The experimental UV-Visible spectrum is recorded in the DMSO solvent and depicted in Figure 6C. From the computed UV-Visible analysis, it can be concluded here that the DMSO solvent has a blueshift effect on the absorption wavelength of the title compound. The bandgap in the DMSO solvent is higher than the gas phase UV-Visible spectrum. The theoretical absorption wavelength in the DMSO solvent is 371.88 nm and the experimental absorption wavelength in the DMSO solvent is 382.07 nm. There is good agreement between the theoretical UV-Visible spectrum (DMSO) and the experimental UV-Visible spectrum (DMSO).
Table 4: Absorption energies (λ in nm), oscillator strength (ƒ), and transitions computed at TD-DFT B3LYP/6-311++G (d,p) level of theory and experimental UV-Visible wavelength (given in bracket and made bold)
|
Gas phase
|
DMSO
|
|
Config
|
Coefficient
|
f
|
λ, nm
|
Config
|
Coefficient
|
f
|
λ, nm
|
|
103 ->104
|
0.69812
|
0.4625
|
372.49
|
103 ->104
|
0.69414
|
0.3611
|
371.88
(382.07)
|
Config – Configuration
Vibrational Assignments and Thermochemical Study
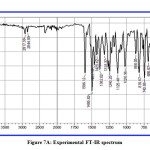
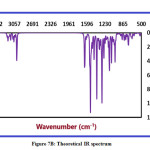
The theoretical and experimental IR spectra of CPMPP molecule are represented in Figure 7A and 7B respectively. The comparative investigations between the scaled theoretical and experimental IR frequencies are tabulated in Table 5. The total numbers of atoms in the titled molecule are 49 and therefore it will have 141 fundamental modes of vibrations. The point group symmetry is C1. The DFT computed IR spectrum overestimates the vibrational frequencies and therefore a scaling factor of 0.9613 has been used to correct the vibrational assignments. The results obtained in the theoretical results are showing good agreement with those obtained in experimental data.
Table 5: Selected experimental and theoretical vibrational assignments of studied compound calculated at B3LYP/6-311++ G (d,p) level
|
Mode
|
Computed scaled frequencies (cm-1)
|
IR Intensity (km) mol-1
|
Experimental frequencies (cm-1)
|
Assignment
|
|
75
|
1057
|
69.76
|
1020
|
Ar-Cl str
|
|
91
|
1242
|
244.74
|
1240
|
Ring C Ar-H ip bend
|
|
98
|
1307
|
12.75
|
–
|
Ring A Ar-H ip bend
|
|
100
|
1343
|
318.17
|
1318
|
C9-N7 str
|
|
101
|
1381
|
14.73
|
1382
|
C3-H6 ip bend
|
|
106
|
1430
|
10.22
|
1425
|
C41-H42, C41-H44 ip bend
|
|
113
|
1479
|
103.56
|
1495
|
Ring C aro C=C str
|
|
115
|
1549
|
17.72
|
–
|
Ring A aro C=C str
|
|
116
|
1551
|
2.50
|
–
|
Ring B aro C=C str
|
|
120
|
1583
|
41.79
|
1596
|
C=N str
|
|
122
|
2891
|
24.92
|
2844
|
C3-H6 str
|
|
130
|
3036
|
8.15
|
2917
|
C30-H33 str
|
|
131
|
3039
|
4.84
|
–
|
C14-H18, C16-H19, C12-H17 str
|
|
138
|
3081
|
4.70
|
–
|
Ring C Ar-H str
|
Abbreviations: Ar-aryl, str-stretching, ip bend- in plane bending, aro- aromatic, Ring A: phenyl ring, Ring B: 4-chlorophenyl ring, Ring C: 3,4-dimethoxyphenyl ring.
Various thermodynamic functions have been obtained from the harmonic vibrational frequencies and are presented in the Table 6. The thermochemical parameters like Total energy (thermal), total molar heat capacity (Cv), total entropy (S), zero-point vibrational energy, and rotational constants have been established. Importantly, on the basis of the data provided in this, one can estimate other thermodynamic function with the help of their thermodynamic relationships.
Table 6: Thermochemical parameters by DFT/B3LYP with 6-311++G (d,p) basis set
|
Parameters
|
Value
|
|
Total E Thermal kcal/mol
|
259.79
|
|
Translational
|
0.889
|
|
Rotational
|
0.889
|
|
Vibrational
|
258.01
|
|
Total Molar capacity at constant volume (Cv) Cal mol-1Kelvin-1
|
95.52
|
|
Translational
|
2.98
|
|
Rotational
|
2.98
|
|
Vibrational
|
89.56
|
|
Total entropy (S) Cal mol-1Kelvin-1
|
177.67
|
|
Translational
|
43.79
|
|
Rotational
|
36.99
|
|
Vibrational
|
96.897
|
|
Zero-point vibrational energy (kcal/mol)
|
244.14
|
|
Rotational Constant (GHz)
|
0.21726
|
|
|
0.07687
|
|
|
0.06175
|
|
E (RB3LYP) (a.u.)
|
-1609.51
|
|
Dipole moment (Debye)
|
4.59
|
Conclusion
In summary, pyrazoline derivative is synthesized from chalcones. For a detailed structural examination, spectroscopic, and quantum chemical analysis density functional theory (DFT) method with a basis set 6-311++G (d,p) has been employed. The geometry of the molecule was optimized by using a 6-311++G (d,p) basis set and the geometrical parameters like bond lengths and bond angles have been computed at the same level of theory. The frontier molecular orbital study has been effectively presented to analyze the chemical reactivity of the molecule. By using HOMO-LUMO energies various electron and quantum chemical parameters have been established. Absorption energies, oscillator strength, and electronic excitations have been calculated at TD-6-311++G (d,p) level of theory for 6-311++G (d,p) optimized geometries. The UV-Visible analysis suggests that there is good agreement between the theoretical UV-Visible spectrum (DMSO) and the experimental one. The C2 carbon atom is the most electronegative whereas C20 carbon atom is the most electropositive carbon atom. The scaled theoretical vibrational bands have been compared with the experimental frequencies and a good deal of agreement has found.
Acknowledgments
Authors acknowledge central instrumentation facility (CIF), Savitribai Phule Pune University, Pune for NMR, MGV’s Pharmacy College, Nashik for UV-Visible, and central instrumentation centre (CIC), KTHM College, Nashik for FT-IR spectral analysis. Authors would like to express their sincere and humble gratitude to Prof. Arun B. Sawant for Gaussian study. Dr. Aapoorva P. Hiray, Coordinator, MG Vidyamandir institute, Nashik is gratefully acknowledged for Gaussian package.
Funding Source
We have not received any kind of fund for the research work.
Conflict of Interest
Authors declared that he do not have any conflict of interest regarding this research article.
References
- Alam, M.J., Alam, O., Alam, P. and Naim, M.J., 2015. A review on pyrazole chemical entity and biological activity. International Journal of Pharmaceutical Sciences and Research, 6, pp.1433-1442.
- Chauhan, A., Sharma, P.K. and Kaushik, N., 2011. Pyrazole: a versatile moiety. International Journal of ChemTech Research, 3(1), pp.11-17.
- Karrouchi, K., Radi, S., Ramli, Y., Taoufik, J., Mabkhot, Y.N. and Al-Aizari, F.A., 2018. Synthesis and pharmacological activities of pyrazole derivatives: a review. Molecules, 23(1), p.134.
- Balbi, A., Anzaldi, M., Macciò, C., Aiello, C., Mazzei, M., Gangemi, R., Castagnola, P., Miele, M., Rosano, C. and Viale, M., 2011. Synthesis and biological evaluation of novel pyrazole derivatives with anticancer activity. European Journal of Medicinal Chemistry, 46(11), pp.5293-5309.
- Xu, Z., Gao, C., Ren, Q.C., Song, X.F., Feng, L.S. and Lv, Z.S., 2017. Recent advances of pyrazole-containing derivatives as anti-tubercular agents. European Journal of Medicinal Chemistry, 139, pp.429-440.
- Rahimizadeh, M., Pordel, M., Bakavoli, M., Rezaeian, S. and Sadeghian, A., 2010. Synthesis and antibacterial activity of some new derivatives of pyrazole. World Journal of Microbiology and Biotechnology, 26(2), pp.317-321.
- Hassan, S.Y., 2013. Synthesis, antibacterial and antifungal activity of some new pyrazoline and pyrazole derivatives. Molecules, 18(3), pp.2683-2711.
- El-Moghazy, S.M., Barsoum, F.F., Abdel-Rahman, H.M. and Marzouk, A.A., 2012. Synthesis and anti-inflammatory activity of some pyrazole derivatives. Medicinal Chemistry Research, 21(8), pp.1722-1733.
- Mironov, M.E., Poltanovich, A.I., Rybalova, T.V., Dolgikh, M.P., Tolstikova, T.G. and Shults, E.E., 2020. Synthesis and analgesic activity of 1, 3, 5-trisubstituted pyrazoles containing a diterpenoid moiety. Russian Chemical Bulletin, 69, pp.537-546.
- Naim, M.J., Alam, O., Farah Nawaz, M., Alam, J. and Alam, P., 2016. Current status of pyrazole and its biological activities. Journal of Pharmacy & Bioallied Sciences, 8(1), p.2.
- López-Viseras, M.E., Fernández, B., Hilfiker, S., González, C.S., González, J.L., Calahorro, A.J., Colacio, E. and Rodríguez-Diéguez, A., 2014. In vivo potential antidiabetic activity of a novel zinc coordination compound based on 3-carboxy-pyrazole. Journal of inorganic biochemistry, 131, pp.64-67.
- Bekhit, A.A., Hymete, A., Asfaw, H. and Bekhit, A.E.D.A., 2012. Synthesis and Biological Evaluation of Some Pyrazole Derivatives as Anti‐Malarial Agents. Archiv der Pharmazie, 345(2), pp.147-154.
- Adole, V.A., Pawar, T.B., Koli, P.B. and Jagdale, B.S., 2019. Exploration of catalytic performance of nano-La2O3 as an efficient catalyst for dihydropyrimidinone/thione synthesis and gas sensing. Journal of Nanostructure in Chemistry, 9(1), pp.61-76.
- Fahim, A.M. and Shalaby, M.A., 2019. Synthesis, biological evaluation, molecular docking and DFT calculations of novel benzenesulfonamide derivatives. Journal of Molecular Structure, 1176, pp.408-421.
- de Marco, B.A., Rechelo, B.S., Tótoli, E.G., Kogawa, A.C. and Salgado, H.R.N., 2019. Evolution of green chemistry and its multidimensional impacts: A review. Saudi pharmaceutical journal, 27(1), pp.1-8.
- Adole, V.A., Pawar, T.B. and Jagdale, B.S., 2020. Aqua‐mediated rapid and benign synthesis of 1,2,6,7‐tetrahydro‐8H‐indeno[5,4‐b]furan‐8‐one‐appended novel 2‐arylidene indanones of pharmacological interest at ambient temperature. Journal of the Chinese Chemical Society, 67(2), pp.306-315.
- Patil, B.N., Sathe, P.A., Parade, B.S., Vadagaonkar, K.S. and Chaskar, A.C., 2018. Transition metal free one pot synthesis of aryl carboxylic acids via dehomologative oxidation of styrenes. Tetrahedron Letters, 59(49), pp.4340-4343.
- Adole, V.A., Jagdale, B.S., Pawar, T.B. and Sagane, A.A., 2020. Ultrasound promoted stereoselective synthesis of 2,3-dihydrobenzofuran appended chalcones at ambient temperature. South African Journal of Chemistry, 73, pp.35-43.
- Patil, B.N., Lade, J.J., Karpe, A.S., Pownthurai, B., Vadagaonkar, K.S., Mohanasrinivasan, V. and Chaskar, A.C., 2019. Transition metal-catalyzed CH functionalization of arylacetic acids for the synthesis of benzothiadiazine 1, 1-dioxides. Tetrahedron Letters, 60(13), pp.891-894.
- Adole, V.A., 2020. Synthetic approaches for the synthesis of dihydropyrimidinones/thiones (Biginelli adducts): a concise review. World Journal of Pharmaceutical Research, 9(6), pp. 1067-1091.
- Adole, V.A., More, R.A., Jagdale, B.S., Pawar, T.B. and Chobe, S.S., 2020. Efficient Synthesis, Antibacterial, Antifungal, Antioxidant and Cytotoxicity Study of 2‐(2‐Hydrazineyl) thiazole Derivatives. ChemistrySelect, 5(9), pp.2778-2786.
- Patnala, H., Abbo, H.S., Potla, K.M., Titinchi, S.J. and Chinnam, S., 2019. Polyethylene glycol (PEG-400): An efficient one-pot green synthesis and anti-viral activity of novel α-diaminophosphonates. Phosphorus, Sulfur, and Silicon and the Related Elements, 194(11), pp.1035-1039.
- Chobe, S.S., Adole, V.A., Deshmukh, K.P., Pawar, T.B. and Jagdale, B.S., 2014. Poly (ethylene glycol)(PEG-400): A green approach towards synthesis of novel pyrazolo [3, 4-d] pyrimidin-6-amines derivatives and their antimicrobial screening. Archives of Applied Science Research, 6(2), pp.61-66.
- Subramanian, N., Sundaraganesan, N. and Jayabharathi, J., 2010. Molecular structure, spectroscopic (FT-IR, FT-Raman, NMR, UV) studies and first-order molecular hyperpolarizabilities of 1, 2-bis (3-methoxy-4-hydroxybenzylidene) hydrazine by density functional method. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 76(2), pp.259-269.
- Adole, V.A., Jagdale, B.S., Pawar, T.B. and Sawant, A.B., 2020. Experimental and theoretical exploration on single crystal, structural, and quantum chemical parameters of (E)‐7‐(arylidene)‐1,2,6,7‐tetrahydro‐8H‐indeno[5,4‐b]furan‐8‐one derivatives: A comparative study. Journal of the Chinese Chemical Society.
- Raja, M., Muhamed, R.R., Muthu, S. and Suresh, M., 2017. Synthesis, spectroscopic (FT-IR, FT-Raman, NMR, UV–Visible), NLO, NBO, HOMO-LUMO, Fukui function and molecular docking study of (E)-1-(5-bromo-2-hydroxybenzylidene) semicarbazide. Journal of Molecular Structure, 1141, pp.284-298.
- Adole, V.A., Waghchaure, R.H., Jagdale, B.S., Pawar, T.B. and Pathade, S.S., Molecular tructure, frontier molecular orbital and spectroscopic examination on dihydropyrimidinones: a comparative computational approach Journal of Advanced Scientific Research, 2020. 11 (2), pp.64-70.
- Raja, M., Muhamed, R.R., Muthu, S., Suresh, M. and Muthu, K., 2017. Synthesis, spectroscopic (FT-IR, FT-Raman, NMR, UV–Visible), Fukui function, antimicrobial and molecular docking study of (E)-1-(3-bromobenzylidene) semicarbazide by DFT method. Journal of Molecular Structure, 1130, pp.374-384.
- Alaşalvar, C., Öztürk, N., Alaa, A.M., Gökce, H., El-Azab, A.S., El-Gendy, M.A. and Sert, Y., 2018. Molecular structure, Hirshfeld surface analysis, spectroscopic (FT-IR, Laser-Raman, UV–vis. and NMR), HOMO-LUMO and NBO investigations on N-(12-amino-9, 10-dihydro-9, 10-ethanoanthracen-11-yl)-4-methylbenzenesulfonamide. Journal of Molecular Structure, 1171, pp.696-705.
- Adole, V.A., Waghchaure, R.H., Jagdale, B.S. and Pawar, T.B., 2020. Investigation of Structural and Spectroscopic Parameters of Ethyl 4-(4-isopropylphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate: a DFT Study. Chemistry & Biology Interface, 10(1).
- Sarojinidevi, K., Subramani, P., Jeeva, M., Sundaraganesan, N., SusaiBoobalan, M. and VenkatesaPrabhu, G., 2019. Synthesis, molecular structure, quantum chemical analysis, spectroscopic and molecular docking studies of N-(Morpholinomethyl) succinimide using DFT method. Journal of Molecular Structure, 1175, pp.609-623.
- Shakila, G., Saleem, H. and Sundaraganesan, N., 2017. FT-IR, FT-Raman, NMR and UV Spectral investigation: Computation of vibrational frequency, chemical shifts and electronic structure calculations of 1-bromo-4-nitrobenzene. World Scientific News, 61(2), pp.150-185.
- Khamees, H.A., Mohammed, Y.H., Swamynayaka, A., Al‐Ostoot, F.H., Sert, Y., Alghamdi, S., Khanum, S.A. and Madegowda, M., 2019. Molecular structure, DFT, vibrational spectra with fluorescence effect, hirshfeld surface, docking simulation and antioxidant activity of thiazole derivative. ChemistrySelect, 4(15), pp.4544-4558.
- Pandey, M., Muthu, S. and Gowda, N.N., 2017. Quantum mechanical and spectroscopic (FT-IR, FT-Raman, 1H, 13C NMR, UV-Vis) studies, NBO, NLO, HOMO, LUMO and Fukui function analysis of 5-Methoxy-1H-benzo [d] imidazole-2 (3H)-thione by DFT studies. Journal of Molecular Structure, 1130, pp.511-521.
- Selvaraj, S., Rajkumar, P., Kesavan, M., Thirunavukkarasu, K., Gunasekaran, S., Devi, N.S. and Kumaresan, S., 2020. Spectroscopic and structural investigations on modafinil by FT-IR, FT-Raman, NMR, UV–Vis and DFT methods. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 224, p.117449.
- Moghanian, H., Mobinikhaledi, A. and Monjezi, R., 2013. Synthesis, spectroscopy (vibrational, NMR and UV–vis) studies, HOMO–LUMO and NBO analysis of 8-formyl-7-hydroxy-4-methylcoumarin by ab initio calculations. Journal of Molecular Structure, 1052, pp.135-145.
- Sadgir NV, Dhonnar SL, Jagdale BS, Sawant AB. Synthesis, spectroscopic characterization, XRD crystal structure, DFT and antimicrobial study of (2E)-3-(2, 6-dichlorophenyl)-1-(4-methoxyphenyl)-prop-2-en-1-one. SN Applied Sciences. 2020 Aug;2(8):1-2.
- Chandralekha, B., Rajagopal, H., Muthu, S. and Sevvanthi, S., 2020. Spectroscopic (FT-IR, FT-RAMAN, NMR, UV–Vis) investigations, computational analysis and molecular docking study of 5-bromo-2-hydroxy pyrimidine. Journal of Molecular Structure, p.128494.
- Murugavel, S., Ravikumar, C., Jaabil, G. and Alagusundaram, P., 2019. Synthesis, crystal structure analysis, spectral investigations (NMR, FT-IR, UV), DFT calculations, ADMET studies, molecular docking and anticancer activity of 2-(1-benzyl-5-methyl-1H-1,2,3-triazol-4-yl)-4-(2-chlorophenyl)-6-methoxypyridine–a novel potent human topoisomerase IIα inhibitor. Journal of Molecular Structure, 1176, pp.729-742.
- Jagdale, B.S. and Pathade, S.S., 2019. Synthesis, Characterization and Theoretical Study of 3-(4-bromophenyl)5-(2,4-dichlorophenyl)-1-phenyl-4,5-dihydro-1H-pyrazole Journal of Applicable Chemistry, 8(1), pp.12-19.
- Hiremath, S.M., Khemalapure, S.S., Hiremath, C.S., Patil, A.S. and Basanagouda, M., 2020. Quantum chemical computational and spectroscopic (IR, Raman, NMR, and UV) studies on the 5-(5-methoxy-benzofuran-3-ylmethyl)-3H-[1,3,4] oxadiazole-2-thione. Journal of Molecular Structure, p.128041.
- Manjusha, P., Muthu, S. and Raajaraman, B.R., 2020. Density functional studies and spectroscopic analysis (FT-IR, FT-Raman, UV–visible, and NMR) with molecular docking approach on an antifibrotic drug Pirfenidone. Journal of Molecular Structure, 1203, p.127394.
- Tagore, S.S., Swaminathan, J., Manikandan, D., Gomathi, S., Ram, S.N., Ramalingam, M. and Sethuraman, V., 2020. Molecular, vibrational (FT-IR and FT-Raman), NMR and UV spectral analysis of imidazo [1, 2-b] pyridazine using experimental and DFT calculations. Chemical Physics Letters, 739, p.136943.
- Khemalapure, S.S., Katti, V.S., Hiremath, C.S., Basanagouda, M., Hiremath, S.M., Armaković, S.J. and Armaković, S., 2019. Molecular structure, optoelectronic properties, spectroscopic (FT-IR, FT-Raman and UV–Vis), H-BDE, NBO and drug likeness investigations on 7, 8-benzocoumarin-4-acetic acid (7BAA). Journal of Molecular Structure, 1195, pp.815-826.
- Aayisha, S., Devi, T.R., Janani, S., Muthu, S., Raja, M. and Sevvanthi, S., 2019. DFT, molecular docking and experimental FT-IR, FT-Raman, NMR inquisitions on “4-chloro-N-(4, 5-dihydro-1H-imidazol-2-yl)-6-methoxy-2-methylpyrimidin-5-amine”: Alpha-2-imidazoline receptor agonist antihypertensive agent. Journal of Molecular Structure, 1186, pp.468-481.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM. Gaussian 03 (Revision E. 01), Gaussian, Inc, Wallingford CT, 2004
Views: 1,213
 This work is licensed under a Creative Commons Attribution 4.0 International License.
This work is licensed under a Creative Commons Attribution 4.0 International License.