Comparison of different oxidation techniques for biofunctionalization of pyrolyzed carbon
Varun Penmatsa1, Hiroshi Kawarada2, Yin Song1, and Chunlei Wang1*
1Department of Mechanical and Materials Engineering, Florida International University. Miami, USA .
2School of Science and Engineering, Waseda University. Tokyo, Japan.
DOI : http://dx.doi.org/10.13005/msri/110101
Article Publishing History
Article Received on : 20 Aug 2014
Article Accepted on : 05 Sept 2014
Article Published : 31 Aug 2014
Plagiarism Check: Yes
Article Metrics
ABSTRACT:
Pyrolyzed carbon micro/nano-structures have great potential as functional units in biosensors where biofunctionalization of the carbon surface is a requisite. In this work, we present a comparison of four different oxidation pretreatments, i.e. vacuum ultraviolet (VUV), electrochemical activation (EA), oxygen reactive ion etching (RIE), and ultraviolet/ozone (UV/O3) pretreatments on pyrolyzed carbon surface. X-ray photoelectron spectroscopy (XPS) results indicated that all the oxidation techniques except UV/O3 pretreatment yielded identical oxidation levels. The percentage of the carboxyl group which is suitable for covalent attachment of amine terminated biomolecules increased with pretreatment time, and was highest in the case of VUV pretreatment (15%) followed by oxygen RIE (12.5%) and EA pretreatments (12.5%) and UV/O3 pretreatment showed significantly lower carboxyl group percentage at 6%. This study helps to optimize the surface functionalization conditions for covalent binding of bioreceptors on the pyrolyzed carbon substrate for biosensing applications.
KEYWORDS:
Carbon-MEMS, pyrolyzed carbon, functionalization, vacuum ultraviolet, electrochemical activation, oxygen reactive ion etching, ultraviolet/ozone
Copy the following to cite this article:
Penmatsa V, Kawarada H, Song Y, Wang C. Comparison of different oxidation techniques for biofunctionalization of pyrolyzed carbon. Mat.Sci.Res.India;11(1)
|
Copy the following to cite this URL:
Penmatsa V, Kawarada H, Song Y, Wang C. Comparison of different oxidation techniques for biofunctionalization of pyrolyzed carbon. Mat.Sci.Res.India;11(1). Available from: http://www.materialsciencejournal.org/?p=510
|
Introduction
To improve the stability and detection performance of biosensors it is important to find suitable substrates for conjugating target specific bioreceptors and develop strong attachment chemistry [1]. An ideal substrate should be chemically robust, easily functionalized, and compatible with a wide variety of analytical modalities (fluorescence, surface plasmon resonance, microscopy, electrochemistry, etc.). Recently, many efforts have been invested on the development of micro/nano-electrode architectures using nanotechnology and MEMS techniques, in order to develop highly sensitive biosensing devices [2, 3]. Therefore, parallel with the development of miniaturized electrodes, considerable attention is also being concentrated on studying various nanoelectrode/bio interfaces.
Glass, silicon and gold are commonly used biosensor substrates due to their well-defined functionalization methods, for example: the silanization technique for glass and silicon, [4,5] and thiolization for gold [6]. On the other hand, carbon-based materials are considered attractive alternatives since they offer good electrical conductivity, better resistance towards biofouling and superior stability over conventional substrates when exposed to prolonged incubations in aqueous solutions at elevated temperatures and/or serial hybridizations [7,8]. Another key advantage of using carbon-based materials is that their surfaces can be easily modified using physical, chemical or electrochemical techniques.
In the recent past, microfabrication of carbonaceous material using the Carbon-MEMS process, which is based on the pyrolysis of patterned organic photoresist polymers under inert ambience, has been used in biosensing devices, electrochemical sensors and miniaturized energy storage/energy conversion devices [9-18]. This technology has great potential to extend the practical application of MEMS in biosensing. There have been recent reports on the application of pyrolyzed carbon microelectrodes in different biosensors, including glucose, PDGF-BB and DNA sensing with notable results [19-22]. In essence, to take advantage of the benefits of pyrolyzed carbon in biosensors, the surface of carbon needs to be properly functionalized with chemical groups such as: carboxylic group, amine group, sulfhydryl group, and hydroxyl group to attach biologically derived materials such as recombinant antibodies, engineered proteins, aptamers etc. for the detection of wide variety of physiological substances.
Among different functional groups, the carboxyl group (-COOH) formed as a result of the oxidation of the carbon surface is widely used to bind covalently with amine-terminated biomolecules via amide bonding. Many different oxidation techniques have been used for functionalizing the carbon surface, such as wet chemical techniques [23] oxygen-plasma or atom-beam treatments [24], hot-filament techniques [25], thermal oxidation in oxygen atmosphere [26,27], photochemical procedures, such as ozone exposure [28] and electrochemical oxidation [29,30]. However, it is often quite difficult to compare the different oxidation methods due to the different analysis techniques, setups and evaluation procedures used in various studies. Furthermore, the result of the oxidation process (with respect to both the amount of oxygen as well as type of carbon–oxygen groups) may also depend on the nature of the carbon surface. The functional groups are typically quantified by X-Ray photoelectron spectroscopy (XPS) spectra and to a lesser degree by fourier transform infrared spectroscopy (FTIR).
In this work, we present a comparison of four oxidation pretreatment techniques on pyrolyzed carbon substrates. Information on the various carbon-oxygen groups present on the surface was deduced by analysis of the binding energies of different chemical groups from the deconvoluted C1s core level. Specifically, we will focus on the following questions: (i) Do the different oxidation techniques yield different amounts of adsorbed oxygen?; (ii) Which types of carbon-oxygen groups are found on the surface and do they differ between the oxidation techniques? All the oxidation techniques except UV/O3 pretreatment yielded higher oxidation levels. It was observed that different oxygen-containing groups coexisted and the percentage of carboxyl groups was higher in the case of VUV, oxygen RIE and EA pretreatments compared to UV/O3 pretreatment.
Materials and Methods:
Pyrolyzed Carbon Films Fabrication
The pyrolyzed carbon films were prepared via a typical C-MEMS process [31]. SU-8 25, a negative-tone photoresist was spin coated in a typical process using Headway research™ photoresist spinner at 500 rpm for 12 s then 3000 rpm for 30 s on silicon oxide wafers (4 in. size, (1 0 0)-oriented, n-type). Following the spin coating process, the photoresist was soft baked at 65 °C for 3 min and hard baked at 95 °C for 7 min on a hot plate. To crosslink polymer chains in the photoresist, it was then exposed by a broadband mercury lamp for 20 s using an OAI Hybralign contact aligner (light intensity, 12 mW/cm2). Post expose bake was carried out at 65 °C for 1 min and 95 °C for 3 min to further harden the crosslinked photoresist. To carbonize the samples, photoresist films were heated at a 5 °C/min rate from room temperature to 900 °C and held for 60 min and then cooled down to room temperature. The whole carbonization process was conducted in forming gas (95% N2 + 5% H2) atmosphere with constant gas flow rate at 500 sccm. It should however be noted that unpatterned pyrolyzed carbon films were used in this work for the ease of getting XPS spectra.
Vacuum Ultraviolet (VUV) Surface Treatment
The VUV system used in this work applied a xenon excimer (Xe) lamp to generate an ultraviolet (VUV) light with a central wavelength of 172 nm. After the sample was inserted, the chamber was evacuated and oxygen gas (O2) was introducedtill it reached the required pressure of 3.0 × 104 Pa. VUV treatment was conducted at 20 W of lamp power and light intensity of 12 mW/cm2 for durations varying from 15-120 min. The whole treatment process was done at room temperature. The excimer light was transmitted through the glass window of the lamp housing and chemical reaction chains were triggered in the irradiation chamber. The working principle of the VUV surface treatment is extensively discussed [32]. Up to date there are no reports to the best of our knowledge which can demonstrate VUV pretreatment for the biofunctionalization of pyrolyzed carbon surface.
Electrochemical Activation
To perform the electrochemical activation, the C-MEMS electrodes were connected with a piece of copper wire. Then the contact pad and the silver wire were fully covered by epoxy resin to prevent their exposure to the electrolyte. Subsequently, the sample was configured as the working electrode in a three-electrode system. The reference and the counter electrodes used were Ag/AgCl and a Pt wire, respectively. The activation process was performed in 0.5 M H2SO4 solution deaerated by nitrogen bubbling for time intervals between 5-30 min. A voltage of 1.9 V was applied to the electrodes for the durations ranging from 10-30 min using a multichannel potentiostat/galvanostat (VMP3, Princeton Applied Research). The electrodes were then negatively polarized at -0.3 V for 10 min. After electrochemical pretreatment, the electrodes were washed with DI water.
UV/Ozone (O3) treatment
The ultraviolet rays wavelengths radiated from a low-pressure mercury vapor lamp are 184.9 nm and 253.7 nm. The UV/ozone pretreatment was performed by using UV ozone cleaner UVy253 (Nippon Laser and Electronics Laboratory). At first, the reaction chamber was purged with nitrogen gas for 5 min to remove any active gases. Subsequently oxygen (O2) gas is introduced for 5 min. After turning of the oxygen gas supply, the UV light was turned on for times ranging from 10-180 min. Finally, after turning off the UV source, nitrogen gas was introduced for 5 min to purge the ozone in the reaction chamber before opening the chamber door.
Oxygen RIE pretreatment
MARCH CS-1217 RIE system was used to treat the pyrolyzed carbon surface with oxygen plasma. This system has parallel plate reactor equipped with 13.65 MHz RF source. The gas line for oxygen was completely evacuated before the process to remove any moisture. The oxygen RIE time was varied from 1-10 min.
XPS Analysis
The XPS analysis was investigated by an Ulvac Φ 3300 XPS (Ulvac-Phi) with an anode source providing Al Kα radiation. The electron takeoff angle was 45 ± 3° relative to the substrate surface and the C1s peak was chosen as the reference binding energy (284.6 eV). Spectrum fitting routine was done with the following constraints: Shirley background was used. All peak contributions are mixed Gauss–Lorentz product functions with identical shapes and widths.
Discussion
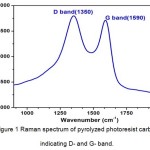
For fabricating pyrolyzed photoresist carbon using the C-MEMS strategy, the photoresist polymer is patterned by conventional photolithography and subsequently carbonized at high temperature under inert atmosphere. It is interesting to note that by changing the lithography conditions, pyrolysis temperature, time, and atmosphere, this versatile technique permits a wide variety of interesting MEMS applications that employ structures with a wide variety of sizes, shapes, electrical and mechanical properties. Raman spectrum of the pyrolyzed photoresist carbon prepared by the C-MEMS technique is shown in Figure 1. The spectrum indicates two broad peaks with one around 1590 cm−1 (G-band) and another around 1325 cm−1 (D-band). The ID/IG ratio of ~1 and the broad D- and G-peaks indicate that photoresist carbon pyrolyzed at 900 °C, has disorder or amorphous phase similar to conventional glassy carbon pyrolyzed at the identical temperature [33].
Figure. 2 shows the typical X-ray photoelectron spectroscopy broad scan spectra of bare carbon, VUV, EA, oxygen RIE and UV/O3 pretreated carbon. The four oxidation pretreatment techniques were chosen based on their suitability towards pyrolyzed carbon films. For example, typical wet oxidation technique using HNO3/H2SO4 mixture is very harsh and thus peels off the carbon film from the substrate. The small peak in the XPS spectrum for bare carbon represents the native surface carbon-oxygen bonds. After oxidation with pretreatments, it can be observed that the oxygen peak is more apparent and is highest in the case of VUV functionalization. This indicates preeminent surface oxidation using VUV pretreatment compared to the other pretreatment techniques. The measured oxygen content which quantifies the surface oxidation is calculated from these broad scan XPS spectra by dividing the peak intensity of oxygen with peak intensity of carbon. The peak intensity is determined by integrating the area under the corresponding peak in the XPS spectrum.
The summary of measured oxygen content as a function of oxidation time in the case of VUV, EA, oxygen RIE and UV/O3 pretreatment techniques is shown in Figure 3. Analysis of the graph shows that the achievable oxidation levels on carbon surface were more than 20 at.% in the case of VUV treatment (≈ 24 at.%), EA treatment (≈ 22 at.%), and oxygen RIE pretreatment ( slightly above 20 at.%). However, in the case of UV/ O3 pretreatment, only ≈ 15 at.% oxygen content was achieved. The lower oxidation levels yielded from UV/O3 pretreatment could be possibly due to a comparatively low intensity of the UV-source. It is observed that the oxidation levels saturate after treatment for longer duration in the case of VUV and UV/O3 pretreatments compared to EA and oxygen RIE pretreatments. These results are consistent with the fact that VUV and UV/O3 are milder oxidation techniques which only show a minor increase in the surface roughness after treatment. Alternatively, in the case of both EA and oxygen RIE, as the pretreatment time was prolonged, the significant increase in the surface porosity and surface roughness was observed (results not shown). It is noteworthy that as the EA and oxygen RIE pretreatment time increased over 30 min and 10 min, respectively, the films started to peel/etch from the substrate and the results were not reproducible. The achieved level of oxidation cannot be increased further without completely destroying the carbon films. Thus, we conclude that with respect to the achievable oxidation level, VUV-, EA- and oxygen RIE-techniques yield much higher oxygen concentrations compared to UV/O3 pretreatment. However, this does not necessarily indicate different oxidation behavior of the latter technique. It may also be from a comparatively low UV-intensity, which could yield higher oxygen coverage but only for impracticably long exposure time. As previously mentioned, a significant difference in the surface morphology can be observed based on the chosen oxidation technique. This signifies the fact that the pretreatment technique should be chosen based on the final application of the electrodes and how the surface morphology affects the device performance.
Figure 4 (a-d) shows the high resolution C1s XPS spectra of pyrolyzed carbon surface treated by all the four oxidation techniques. It should be noted that the specified levels of the oxygen concentration obtained by XPS apply to a fictitious homogeneous mixture of the constituents (C and O) over the whole information depth of the photoelectrons. In order to obtain information on the chemical groups present on the surface, further investigation of the C1s core level chemical shifts was conducted. Due to the rather broad individual contributions (FWHM typically 1 eV) the C1s peak was decomposed into various components. The following 5 peaks were deconvoluted: C-C (sp2) at 284.6 eV, C-C (sp3) at 285.3 eV, C+ I (C-O) at 286.2 eV, C+ II (C=O) at 287.6 eV and C+ III (O-C=O) at 289.1 eV [34]. As discussed earlier, the primary functional group we focus on in this study is O=C-OH which can be used to bind covalently with amine-terminated biomolecules. The peak of carboxyl group is stronger in VUV, oxygen RIE, and EA treatments compared to UV/O3 treatments. Another noteworthy observation is that C-C (sp3) peak representing ordered carbon structure is smallest in the oxygen RIE and EA treatments because as discussed previously they alter the surface morphology significantly by introducing porosity and surface roughness.
Figure 5 shows the development of the C-C (sp2 & sp3) and the oxygen-related C1s components with increasing oxidation time on the carbon surface. One common trend that can be observed is that upon oxidation the sp2 contents decrease steadily while the oxygen-related components gain in intensity. For all exposures several oxygen-related species are observed with the C+I component always being the dominating one. Commonly, the C+II component is attributed to carbonyl groups (C=O). However, in the simplest approximation, when final-state effects in XPS are neglected, the chemical shift is directly related to the oxidation state. Thus C+II could as well be caused by chains of bridge-bonded oxygen atoms (i.e. multiple ether groups (―C―O―C―O―C―O―) [34]. Accordingly, rather than signifying a gradual conversion from bridge-bonded ether-like oxygen to on-top “carbonyl” oxygen, the increase of C+II may also be interpreted as formation of chains of ether- like groups, as they are favored by theory [35,36]. It was observed that in most cases the component C+III (representing the carboxyl group) increased with treatment time in all the oxidation techniques. The coverage of the carboxyl group on the surface reached close to 15% in the case of VUV, ≈6% for UV/O3 and ≈12.5% for both EA and oxygen RIE. These values are comparable or better than the coverage values obtained for other oxidation pretreatments on the pyrolyzed carbon surface [23-30]. Finally, it should be noted that to maximize the coverage of functional groups on the carbon surface it is important to fine tune the parameters during the functionalization process. As previously observed, different oxidation techniques produce different effect on the surface morphologies. It is important to select the functionalization technique based on the final application. For example, using oxygen RIE and EA introduces surface porosity and increases effective surface area along with simultaneously oxidizing the surface which could be useful in electrochemical devices and biosensors. On the other hand, VUV and UV/O3 pretreatment techniques are more applicable for applications which require smooth oxidized electrode surface.
Oxidation of pyrolyzed photoresist carbon films was investigated by XPS. The VUV, EA and RIE pretreatments yielded ≥20% oxidation levels while UV/O3 pretreatment only showed 15% surface oxidation. Different oxygen-containing groups coexisted on the carbon surface and the percentage of the carboxyl group was the highest in VUV at 15% followed by oxygen RIE and EA at 12.5% and 6% in the case of UV/O3. This study helps to optimize the surface functionalization technique for covalent binding of bioreceptors on the pyrolyzed carbon substrate. This study helps in the selection and optimization of oxidation techniques for functional groups grafting that are conductive for covalent binding of bioreceptors. These oxidation techniques can potentially be used for the surface functionalization of other carbon surfaces, such as carbon nanotubes, graphene, diamond and glassy carbon.
Acknowledgements:
Varun Penmatsa gratefully acknowledges the financial support from DEA and DYF fellowships provided by FIU graduate school. This project is partially supported by National Science Foundation (OISE 0934078 and CMMI 0800525). The authors would also like to acknowledge Grant-in-Aid from GCOE Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan and a Grant-in-Aid for Fundamental Research A (23246069) of Japan Society for the Promotion of Science (JSPS). The authors would also like to acknowledge the research facilities provided at AMERI at FIU and NTRC at Waseda University.
References:
- Lechleitner T., Klauser F., Seppi T., Lechner J., Jennings P., Perco P., Mayer B., Steinmuller-Nethl D., Preiner J., Hinterdorfer P., Hermann M., Bertel E., Pfaller K. and Pfaller W., Biomaterials, 29, 4275 (2008).
CrossRef
- Penmatsa V., Functionalized carbon micro/nanostructures for biomolecular detection, Florida International University, PhD Thesis, (2012).
- Finot E., Bourillot E., Meunier-Prest R., Lacroute Y., Legay G., Cherkaoui-Malki M., Latruffe N., Siri O., Braunstein P. and Dereux A., Ultramicroscopy, 97, 441 (2003).
CrossRef
- Sarath V., Kumar M., Karanth N. and Thakur M., Biosens Bioelectron., 19, 1337 (2004).
CrossRef
- Song M., Yun D., Min N. and Hong S., J Biosci Bioeng., 103, 32 (2007).
CrossRef
- Dong S. and Li J., Bioelectroch Bioener., 42, 7 (1997).
CrossRef
- Yang W., Auciello O., Butler J., Cai W., Carlisle J., Gerbi J., Gruen D., Knickerbocker T., Lasseter T., Russell J., Smith L. and Hamers R., Nature Materials, 1, 253 (2002).
CrossRef
- Ruslinda A., Penmatsa V., Ishii Y., Tajima S. and Kawarada H., Analyst., 137, 1692 (2012).
CrossRef
- Penmatsa V., Kim T., Beidaghi M., Kawarada H., Gu L., Wang Z. and Wang C., Nanoscale, 4, 3673 (2012).
CrossRef
- Penmatsa V., Yang J., Yu Y. and Wang C., Carbon, 48, 4109 (2010).
CrossRef
- Lee J., Hwang S., Kwak J., Park S., Lee S. and Lee K., Sensor Actuator B, 129, 372 (2008).
CrossRef
- Chen W., Beidaghi M., Penmatsa V., Bechtold K., Kumari L., Li W. and Wang C., Nanotechnology, IEEE Transact, 9, 734 (2010).
- Lee J., Hwang S., Kwak J., Park S., Lee S. and Lee K.. Sensors and Actuat. B: Chem., 129, 372 (2008).
CrossRef
- Kamath R. and Madou M., Anal Chem., 86, 5991 (2014).
CrossRef
- Heo J., Shim D., Teixidor T., Oh S. and Madou M., J Electrochem Soc., 158, J76 (2011).
CrossRef
- Parikh Y., Penmatsa V., Yang J. and Wang C., Proceedings of the COMSOL Conf. Boston, MA, USA (2008).
- Teixidor G., Zaouk R., Park B. and Madou M., J Power Sources, 183, 730 (2008).
CrossRef
- Song Y., Penmatsa V. and Wang C., Energies, 7, 4694 (2014).
CrossRef
- Penmatsa V., Ruslinda A., Beidaghi M., Kawarada H. and Wang C., Biosens Bioelectron., 39, 118 (2013).
CrossRef
- Penmatsa V., Ruslinda A., Beidaghi M., Kawarada H. and Wang C., ECS Trans., 45, 7 [2013].
- Yang J., Penmatsa V., Tajima S., Beidaghi M., Kawarada H. and Wang C., Mater Lett., 63, 2680 (2009).
CrossRef
- Xu H., Malladi K., Wang C., Kulinsky L., Song M. and Madou M., Biosens Bioelectron., 23, 1637 (2008).
CrossRef
- Wang X., Ruslind R. Ishiyama Y., Ishii Y. and Kawarada H., Diam Relat Mater., 20, 1319 (2010).
CrossRef
- Wang W., Huang B., Wang L. and Ye D., Surf. Coat. Tech., 205, 4896 (2001).
CrossRef
- Pehrsson P. and Mercer T., Surf Sci., 460, 74 (2000).
CrossRef
- Ferro S., Colle M. and Battisti A., Carbon, 439, 1191 (2005).
CrossRef
- Nakamura J. and Ito T., Appl Surf Sci., 244, 301 (2005).
CrossRef
- Boukherroub R., Wallart X., Szunerits S., Marcus B., Bouvire P. and Mermoux M., Electrochem comm., 7, 937 (1997).
CrossRef
- Goeting C., Marken F., Gutierrez-Sosa A., Compton R. and Foord J., Diam Relat Mater., 9, 390 (2000).
CrossRef
- Notsu H., Yagi I., Tatsuma T., Tryk D. and Fujishima A., Electrochem. Solid-State Lett., 2, 522 (1999).
CrossRef
- Wang C., Jia G., Taherabadi L. and Madou M., J Microelectromech Syst., 14, 348 (2005).
CrossRef
- Sakuma K., Nagai N., Mizuno J. and Shoji S., Proceedings Electronic Components and Technology Conference. San Diego (CA. USA), 614, (2009).
- Penmatsa V., Kawarada H. and Wang C., J Micromech Microeng., 22, 045024, (2012).
CrossRef
- Strobel P., Ristein J. and Ley L., Diam Relat Mater., 17, 1362 (2008).
CrossRef
- Sque S., Jones R. and Briddon P., Phys Rev B, 73, 085313 (2006).
CrossRef
- Rutter M. and Robertson J., Phys Rev B, 57, 9241 (1998).
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.