Vishnu A. Adole1* and Ganesh B. Yelmame2
and Ganesh B. Yelmame2
1Department of Chemistry, Mahatma Gandhi Vidyamandir’s Arts, Science and Commerce College, (Affiliated to SP Pune University, Pune) Manmad, Nashik-423 104, India.
2Department of Chemistry, Mahatma Gandhi Vidyamandir’s SPH Arts, Science and Commerce College, (Affiliated to Savitribai Phule Pune University, Pune) Nampur, Nashik- 423204, India.
Corresponding Author E-mail: vishnuadole86@gmail.com
DOI : http://dx.doi.org/10.13005/msri/180109
Article Publishing History
Article Received on : 03-Mar-2020
Article Accepted on : 12-Apr-2021
Article Published : 14 Apr 2021
Plagiarism Check: Yes
Reviewed by: Dr. Guru Swamy
Second Review by: Dr. Murugesh V
Final Approval by: Dr. Pawan Tambade
Article Metrics
ABSTRACT:
In the present study, 2-(p-tolyl)-2,3-dihydro-1H-perimidine (TDHP) is synthesized from 1,8-naphthalenediamine and 4-methylbenzaldehyde by embedding a one-carbon unit between the nitrogen followed by ring closure using green chemistry approach. 1H NMR and 13C NMR spectral techniques were used to validate the structure of the TDHP. The synthesized perimidine TDHP is studied using density functional theory (DFT) to provide valuable insights into structural, chemical, and thermochemical study.The structural and chemical properties of TDHP were computed using the DFT method on the B3LYP/6-311G(d,p) basis package. Bond lengths were predicted from the optimised molecular structure, and the physical and chemical properties of the molecules were inferred as a consequence. The HOMO and LUMO are computed, and quantum chemical parameters are determined using electronic energies. The calculated HOMO-LUMO energy gap is 4.25 eV indicating charge transfer phenomenon within the molecule. The electron density and chemical behaviour of the TDHP was predicted using Mulliken atomic charges and the molecular electrostatic surface potential plot.Amongst all carbon atoms, the C8 carbon as more positive and C27 as more negative carbon atoms. The high global electrophilicity index suggests electrophilic character of the TDHP.The harmonic vibrational frequencies were used to measure total energy, total molar entropy, and molar heat capacity.
KEYWORDS:
B3LYP/6-311G(d,p); 3-dihydro-1H-perimidine; DFT; FMO; MESP; 2-(p-Tolyl)-2
Copy the following to cite this article:
Adole V. A, Yelmame G. B. Synthesis and Theoretical Calculations of 2-(p-Tolyl)-2,3-Dihydro-1H-Perimidineusing Density Functional Theory. Mat. Sci. Res. India;18(1).
|
Copy the following to cite this URL:
Adole V. A, Yelmame G. B. Synthesis and Theoretical Calculations of 2-(p-Tolyl)-2,3-Dihydro-1H-Perimidineusing Density Functional Theory. Mat. Sci. Res. India;18(1). Available from: https://bit.ly/3g5OfXz
|
Introduction
Perimidine is a tri-cyclic heterocycle comprising two nitrogen atoms at the first and third positions, which enhances pi-electrons’ delocalization to the naphthalene ring from the fused heterocyclic ring. Perimidines’ biological behaviour has been the topic of numerous journal entries in recent years. Perimidines are robust motifs and an interesting family of N-heterocycles that have advanced significantly in recent years as a result of their wide range of uses in life sciences, medicine, and industrial research. Their way to interact molecularly with protein molecules, form complexes with metal, and behave differently in different light wavelengths renders themselves increasingly enticing for potential use.Perimidines are of great interest to researchers because of their broad variety of biological activities 1,2. The biological profile of several perimidine hybrids is found to be explored in numerous applications such as Pd(II) complexes derived from perimidine ligand for in-vitro antimicrobial applications3, antioxidant properties of transition metal complexes of perimidine ligand 4, anti-inflammatory studies of transition metal complexes of 1H-perimidine derivatives 5, 2-phenylazonaphtho[1,8-ef][1,4] diazepines and 9-(3-arylhydrazono) pyrrolo [1,2-a] perimidines as antitumor agents 6, oxo-7H-benzo [e] perimidine-4-carboxylic acid derivatives as potent, nonpeptide corticotropin releasing factor (CRF) receptor antagonists 7, and 3-Chloro-1-(4-perimidine methylcarbonylamino)-4-phenyl-azetidin-2-one for antibacterial and antifungal activities 8. Besides, 1-alkylperimidineruthenium (II) complexes were used for catalytic applications 9, N-xylyl-N′-methylperimidinecarbene iridium complexes as catalysts for C–H activation and dehydrogenativesilylation 10, Perimidin-2-ylidene rhodium(I) complexes for transfer hydrogenation reaction 11, Perimidine based synthetic receptors for identification of cupric ionin water solution 12,2,3-dihydroperimidine derivaties for lubricants 13, etc.
Theoretical calculations such as DFT has been used to identify various structural, molecular and spectral properties(E)-7-(arylidene)-indanones 14, 2-(3-bromophenyl)-4-(4-bromophenyl)-2,3-dihydro-1H-1,5-benzodiazepine 15, 2-(2-Hydrazineyl) thiazole derivatives 16, (3,5-Diphenyl-4,5-dihydro-1H-pyrazol-1-yl)(phenyl) methanone 17, 2-(5-methyl-1-benzofuran-3-yl) acetic acid 18, 2,4-Dibromoaniline 19, 5-Methoxy-1H-benzo[d]imidazole-2(3H)-thione 20, etc. Besides, DFT methodology has been efficiently used for studying electronic and structural properties of graphyne oxide for CO, CO2 and NH3 adsorption phenomena21, exploration of structural and mechanical properties of pristine and adsorbed puckered arsenene nanostructures 22,investigation of kinetics and mechanism of oxidation of bromothymol blue23, etc.The DFT method with appropriate basis set is found to envisage spectroscopic properties like UV-Visiblein terephthalic acid 24, 7-hydroxy-3(4-methoxyphenyl)chromone 25, bis(thiourea)nickel chloride 26, NMR study inN,N′-bis(salicylidene)-1,2-phenylenediamine 27, 3-fluorophenylboronic acid 28, 5-bromo-2-ethoxyphenylboronic acid (monomer and dimer structures) 29, 5-(3-pyridyl)-4H-1,2,4-triazole-3-thiol 30, veratrole 31, Raman investigation 32-35 and IR assignments’ study in5-(4-chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-pyrazole 36, (E)-1-(2,3-dihydrobenzo [b][1,4]dioxin-6-yl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one 37, dihydropyrimidinones 38, (2E)-3-(2,6-dichlorophenyl)-1-(4-fluoro)-prop-2-en-1-one 39, ethyl-4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 40, indole-3-carboxylic acid 41.
The DFT approach with the B3LYP functional has been shown to predict theoretical properties that are in good agreement with experimental spectroscopic results.42-45. Using the B3LYP functional with a 6-311G(d,p) basis set, the assignment of absorption bands and, as a result, the prediction of electronic and chemical properties of molecules is found to be accurate. 46, 47.Energized by all above mentioned aspects, in the present study, we have synthesized, characterized and studied 2-(p-tolyl)-2,3-dihydro-1H-perimidine using DFT method at B3LYP/6-311G(d,p) basis set.This is first report on DFT study of the TDHP compound. For the TDHPmolecule, no work on DFT analysis for the exploration of structural, electronic, and chemical parameters has been done to date.Structural parameters using optimized molecular structure have been obtained. The optimized structure provides the information of geometrical parameters, stability of the molecule and also polarity. With the help of geometry optimization, HOMO-LUMO energies are computed and these are further used for the determination of electronic and global reactivity parameters. I have predicted the electronic properties such asionization potential energy (I)electron affinity (A) and global reactivity attributes such as hardness (η), softness (S), electronegativity (χ), chemical potential (μ) and electrophilic index (ω) with the help of HOMO-LUMO energies.The band gap energy and frontier molecular orbitals (FMOs) are established and examined. The molecular electrostatic surface potential is established by using optimized structure and being used to locate sites for electrophilic and nucleophilic attacks.
Materials and Methods
General remarks
The 1,8-diamino naphthalene (Purity: 99%, Make: sigma Aldrich) and p-Tolualdehyde (Purity: 98%, Make: Alfa aesar) 98%chemicalswere purchased from Virion Enterprises, Mumbai. The chemicals were used exactly as they were obtained, with no further purification needed.Melting point was determined in open capillary and uncorrected. 1H NMR and 13C NMR spectra were recorded with a Bruker using DMSO-d6solvent on 400 MHz NMR Spectrometer. Thin-layer chromatography using aluminium sheets with silica gel 60 F254 (Merck) was used to track the completion of the reaction.
Synthesis of TDHP
In the past, the p-TSA was employed for the synthesis of 2,3-dihydroquinazolin-4(1H)-one derivatives48. Based on this we tried the synthesis of PDHP using p-TSA using 2 mol% catalyst dose and to our credit, we got good results in terms of synthetic efficiency. We did not switch our attention for optimizing the reaction conditions as our main goal of the present study of structural, electronic and chemical nature of the PDHP molecule.In a standard reaction procedure, a mixture containing naphthalene-1,8-diamine (1, 0.01mmol), p-toluene benzaldehyde (2, 0.01 mmol) and p-TSA (2 mol%) in ethanol (5 mL) were taken in a conical flask and stirred at room temperature for 30 minute on magnetic stirrer.After completion of the reaction (monitored by TLC), the crude product was obtained simply filtration and the it was purified by recrystallization to provide the perimidine derivative (3): 2-(p-tolyl)-2,3-dihydro-1H-perimidine (Scheme 1). The physicochemical and spectral data of title compound is given in Table 1.
Scheme 1: Synthesis of title compound
Table 1: Physicochemical and spectral data of title compound
|
Systematic Name of the Product
|
2-(p-tolyl)-2,3-dihydro-1H-perimidine
|
|
Abbreviation used
|
TDHP
|
|
Physicochemical data
|
Yield: 85 %, M.P: 147 ℃, Colour: Faint brown
|
|
1H NMR
(400 MHz, DMSO)δ
|
9.98 (s, 1H), 8.34 – 8.22 (m, 1H), 7.80 (d, J = 8.3 Hz, 2H), 7.49 – 7.35 (m, 1H), 7.23 (d, J = 8.3 Hz, 2H), 6.99 (dd, J = 7.8, 0.8 Hz, 1H), 6.71 (t, J = 18.7 Hz, 1H), 6.64 (s, 1H), 6.51 (d, J = 8.1 Hz, 1H), 6.51 (d, J = 8.1 Hz, 1H), 4.61 (s, 1H), 2.44 (s, 3H)
|
|
13C NMR
(400 MHz, DMSO) δ
|
143.59, 139.31, 138.18, 134.83, 129.15, 128.25, 127.28, 115.62, 112.92, 104.72, 66.62, 21.27
|
Computational details
DFT measurements of a TDHP molecule were conducted using the Gaussian 03 software package [49]. The molecular structure of the TDHP molecule in the ground state was optimized using the density functional theory (DFT/B3LYP) 50, 51 with the 6-311G(d,p) basis set 52, 53.All of the quantum chemical calculations for the TDHP compound were performed at B3LYP/6-311G(d,p) level and corresponding molecular drawing was done with the aid of the Gauss View 4.1.2 visualization programme 54. Thermochemical data were achieved using harmonic vibrational frequencies.
Results and Discussion
Geometrical Parameters
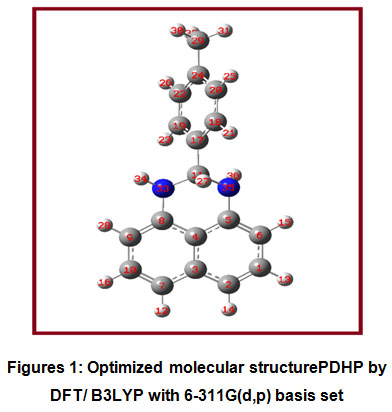
The optimized molecular structure of the title molecule is given in Figure 1. Table 2 shows the optimized geometric parameter bond lengths by B3LYP with the 6-311G(d,p) basis set. The B3LYP method is well known for predicting geometrical parameters that are closer to experimental results, as discussed in the literature 55. B3LYP/6-311G(d,p) is taken into consideration for these purposesin the present discussion.The information about bond lengths is very crucial to get insights into inter and intra molecular interactions, hydrogen bonding effects and also chemical reactivity patterns56-58.For the title compound, the bonds C5-N35 and C8-N33 are having 1.4157 Å and 1.3979 Å respectively. This will makenitrogen atom of C5-N35bond better nucleophile since the bonding electrons are more polarized due to longer bond length.When acidic nature of H34 and H36 is compared, it is observed that H36possessmore acidic strength which is supported by their bond lengths and Mulliken atomic charge values. The bond length values of N33-H34 and N35-H36 bonds are 1.0139 Å and 1.0142 Å respectively. In the naphthalene ring of the TDHP compound, C1-C2 andC7-C10 bonds possess nearly same bond lengths and importantly these two bonds are found to contain maximum double bond character and thereby render electrophilic aromatic substitution reactions more probable at these bonds.The C2 and C9 positions are more prone towards electrophilic attack. In this way the knowledge about bond length values provides valuable insights into physical and chemical properties of the TDHP compound.
Figures 1: Optimized molecular structurePDHP by DFT/ B3LYP with 6-311G(d,p) basis set
Table 2: Optimized geometrical parameters of PDHP by DFT/ B3LYP with 6-311G(d,p) basis set
|
Bond lengths (Å)
|
|
C1-C2
|
1.3788
|
C11-H27
|
1.103
|
|
C1-C6
|
1.4111
|
C11-N33
|
1.4712
|
|
C1-H13
|
1.0864
|
C11-N35
|
1.4571
|
|
C2-C3
|
1.4198
|
C17-C18
|
1.3991
|
|
C2-H14
|
1.0863
|
C17-C19
|
1.399
|
|
C3-C4
|
1.4298
|
C18-C20
|
1.3926
|
|
C3-C7
|
1.4211
|
C18-H21
|
1.0869
|
|
C4-C5
|
1.4253
|
C19-C22
|
1.3953
|
|
C4-C8
|
1.431
|
C19-H23
|
1.0858
|
|
C5-C6
|
1.3839
|
C20-C24
|
1.4022
|
|
C5-N35
|
1.4157
|
C20-H25
|
1.0872
|
|
C6-H15
|
1.0858
|
C22-C24
|
1.3999
|
|
C7-C10
|
1.3776
|
C22-H26
|
1.0872
|
|
C7-H12
|
1.086
|
C24-C29
|
1.5103
|
|
C8-C9
|
1.3853
|
C29-H30
|
1.0971
|
|
C8-N33
|
1.3979
|
C29-H31
|
1.0946
|
|
C9-C10
|
1.4118
|
C29-H32
|
1.0936
|
|
C9-H28
|
1.087
|
N33-H34
|
1.0139
|
|
C10-H16
|
1.0865
|
N35-H36
|
1.0142
|
|
C11-C17
|
1.5149
|
–
|
–
|
Frontier molecular orbital analysis
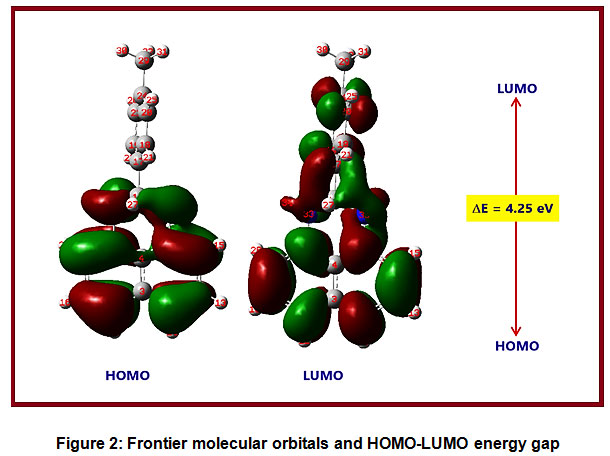
HOMO and LUMO are called as frontier molecular orbitals (FMOs) and very crucial for assessing a molecule’s reactivity and stability. HOMO and LUMO are linked with electron-donating and electron-accepting capabilities respectively. The narrower the energy difference between HOMO and LUMO, the more stable a molecule becomes. A decrease in stability, polarizability, and electron transport in a molecule occurs as the HOMO-LUMO energy gap is increased. Since they relate to ionization potential energy and electron affinity, respectively, the HOMO and LUMO energy values are very important to understand the electronic nature of the molecules.The HOMO-LUMO pictures are given in Figures 2.The HOMO–LUMO energy and reactivity descriptor values of the PDHP Molecule are calculated in the gas phase and presented in Table 3. The HOMO is observed to be principally on naphthalene ring. The HOMO energy in the title molecule is -4.85 eV and LUMO energy is -0.60 eV. When predicting the reactivity pattern between the TDHP compound and other molecules, the HOMO-LUMO values of the TDHP compound could be compared to the HOMO-LUMO values of the other molecules. The estimated band gap value is 4.25 eV. The electron affinity and ionization potential values are 0.60 and 4.85 eV. The global reactivity parameter values are calculated using Koopman’s theorem 59.The global reactivity parameters like chemical hardness (η), chemical softness (S), chemical potential (μ) and electrophilicity index (ω) are calculated using HOMO-LUMO energies. The calculated chemical hardness and chemical softness values are 2.12 eV and 0.47eV-1respectively.These two values are critical in determining the polarizability of the TDHP compound. The existence of intermolecular interactions and chemical reactivity may be predicted by comparison with other molecules. The electrophilicity index value in the title molecule is 1.75 eV.The electrophilicity index is greater than 1.5 eV that indicates the TDHP compound could accept the electrons [58].The chemical potential value is 2.72 eV.
Figure 2: Frontier molecular orbitals and HOMO-LUMO energy gap
Table 3: The HOMO–LUMO energy and reactivity descriptor values of the PDHP Moleculeare calculated in the gas phase
|
Molecular properties
|
Calculated values
|
|
EHOMO energy
|
-4.85 eV
|
|
ELUMO energy
|
-0.60 eV
|
|
ELUMO – EHOMO energy gap (DE)
|
4.25 eV
|
|
Electron affinity (A)
|
0.60 eV
|
|
Ionization energy (I)
|
4.85 eV
|
|
Chemical hardness (η)
|
2.12 eV
|
|
Chemical softness (S)
|
0.47eV-1
|
|
Chemical potential (μ)
|
-2.72 eV
|
|
Electrophilicity index (ω)
|
1.75 eV
|
Mulliken atomic charges
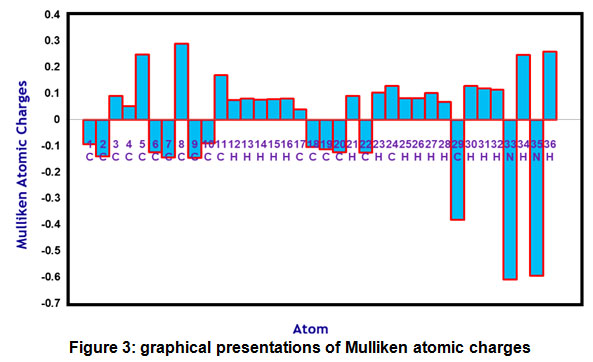
Mulliken charges for the PDHP molecule are determined using the B3LYP/6-311G(d,p) method and are described in Figure 3 and Table 4 to estimate the net atomic populations in the PDHP molecule. Mulliken atomic charges calculation remains essential in the implementation of quantum chemical calculations to molecules because atomic charges impact several attributes of molecules. The two nitrogen atoms of the title molecule are found to be most negative in terms of Mulliken atomic charge with -0.610591 and -0.596207Mulliken atomic charge values for N33 and N35 atoms. The C8 atom is the most positive atom with Mulliken atomic charge value of 0.289643. All hydrogen atoms in the title molecule are positive in terms of Mulliken atom charge.
Figure 3: graphical presentations of Mulliken atomic charges
Table 4: Mulliken atomic charges computed at B3LYP/6-311G(d,p) method
|
Atom
|
Mulliken atomic charge
|
|
1 C
|
-0.093114
|
|
2 C
|
-0.140902
|
|
3 C
|
0.091281
|
|
4 C
|
0.051997
|
|
5 C
|
0.248417
|
|
6 C
|
-0.123987
|
|
7 C
|
-0.144325
|
|
8 C
|
0.289643
|
|
9 C
|
-0.145986
|
|
10 C
|
-0.090983
|
|
11 C
|
0.169852
|
|
12 H
|
0.074204
|
|
13 H
|
0.080591
|
|
14 H
|
0.076251
|
|
15 H
|
0.078505
|
|
16 H
|
0.079317
|
|
17 C
|
0.037742
|
|
18 C
|
-0.104867
|
|
19 C
|
-0.113417
|
|
20 C
|
-0.123714
|
|
21 H
|
0.090465
|
|
22 C
|
-0.125990
|
|
23 H
|
0.103043
|
|
24 C
|
0.128424
|
|
25 H
|
0.081299
|
|
26 H
|
0.081192
|
|
27 H
|
0.100655
|
|
28 H
|
0.067192
|
|
29 C
|
-0.381613
|
|
30 H
|
0.128129
|
|
31 H
|
0.118828
|
|
32 H
|
0.113955
|
|
33 N
|
-0.610591
|
|
34 H
|
0.246391
|
|
35 N
|
-0.596207
|
|
36 H
|
0.258324
|
Molecular electrostatic surface potential and thermochemistry analysis
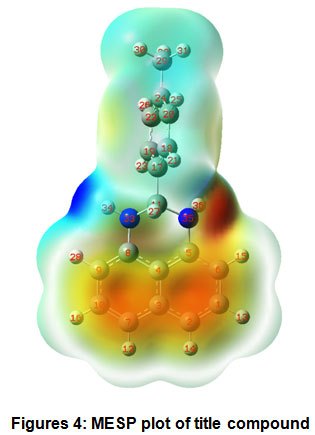
The molecular electrostatic surface potential (MESP) plotof the PDHP molecule given in Figure 4 is used to investigate the chemical reactivity of the title molecule, and it is plotted over the optimized electronic structure of the title compound using density functional B3LYP level with 6-311G(d,p) basis collection. Since it was established in space around a molecule by the charge distribution, the MESP plot is quite valuable in studying the reactive sites for nucleophilic and electrophilic attacks, hydrogen bonding interactions and also in the biorecognition processes. Different colours represent different electrostatic potential values: red symbolizes negative electrostatic potential, blue denotes positive electrostatic potential, and green depicts less positive electrostatic potential. In the title molecule the negative potential is around nitrogen atom and also around naphthalene ring system indicating high electron density which indicatesits reactivity as nucleophile. The highest positive potential is found around hydrogen atoms attached to the nitrogen atoms. The hydrogen atoms are expected to react with bases to form anions. The p-tolyl ring system possesses less positive electrostatic potential values.
Figures 4: MESP plot of title compound
The statistical analysis thermodynamic functions: total thermal energy, heat capacity, and entropy for the PDHP compound were obtained from the theoretical harmonic frequencies and described in Table 5 based on vibrational analysis at the B3LYP/6-311G(d,p) stage.
Table 5: Thermodynamic functions of title compound calculated in the gas phase
|
Entry
|
E (Thermal) KCal/Mol
|
CV
Cal/Mol-Kelvin
|
S
Cal/Mol-Kelvin
|
|
Electronic
|
196.199
|
64.274
|
128.241
|
|
Translational
|
0.000
|
0.000
|
0.000
|
|
Rotational
|
0.889
|
2.981
|
42.568
|
|
Vibrational
|
0.889
|
2.981
|
33.847
|
|
Total
|
194.422
|
58.312
|
33.847
|
Conclusion
The PDHP compound was synthesized using green chemistry approach and1H and 13C NMR spectroscopy was used to affirm the molecular structure of the synthesised compound PDHP. DFT calculations were used to determine the molecular structural parameters, HOMO–LUMO analysis, MESP plot, Mulliken atomic charges, and thermodynamic properties of the title compound for optimized geometry. The geometry was optimised using the DFT/B3LYP/ method with the 6-311G(d,p) basis set without any symmetry constraints.According to the optimized molecular structure and bond length details, C1-C2 and C7-C10 bonds are more vulnerable to electrophilic attack. N35 was revealed to be more reactive to acidic reagent.The two nitrogen atoms in the title molecule are the most negative and on the other hand hydrogen atoms in the title molecule are positivein terms of Mulliken atomic charge.According to MESP, negative potential around the nitrogen atom and even around the naphthalene ring structure in the title molecule shows a high electron density, suggesting its reactivity as a nucleophile. Besides, some thermochemical insights are provided. We conclude that the results of this analysis will allow for further research into the physical and chemical properties of the TDHP and related perimidine derivatives.
Acknowledgment
Authors also would like to thank Principal, Mahatma Gandhi Vidyamandir’s Arts, Science and Commerce College, Manmadfor permission and providing necessary research facilities. Authors would like to express their sincere and humble gratitude to Prof. (Dr.) Arun B. Sawant and Prof. (Dr.) Thansing B. Pawarfor Gaussian study. Dr. Aapoorva P. Hiray, Coordinator, MG Vidyamandir institute, is gratefully acknowledged for Gaussian package.
Conflict of Interest
Authors declared that they have any conflict of interest regarding this research article.
Funding Sourse
We have not received any kind of fund for the research work.
References
- Bu, X., Deady, L.W., Finlay, G.J., Baguley, B.C. and Denny, W.A., 2001. Synthesis and cytotoxic activity of 7-oxo-7 H-dibenz [f, ij] isoquinoline and 7-oxo-7 H-benzo [e] perimidine derivatives. Journal of medicinal chemistry, 44(12), pp.2004-2014.
CrossRef
- Starshikov, N.M. and Pozharskii, F.T., 1973. Synthesis of 2-(5-halogeno-2-f uryl)-2, 3-dihydroperimidines. Chemistry of Heterocyclic Compounds, 9(7), pp.922-924.
CrossRef
- Azam M, Warad I, Al-Resayes SI, Alzaqri N, Khan MR, Pallepogu R, Dwivedi S, Musarrat J, Shakir M (2013) Synthesis and structural characterization of Pd(II) complexes derived from perimidine ligand and their in vitro antimicrobial studies. J Mol Struct 1047(1):48–54
CrossRef
- Syntheses, physic-chemical studies and antioxidant activities of transition metal complexes with a perimidine ligand. AnorgAllgChem 638(5):881–886
CrossRef
- assyouni FA, Abu-Bakr SM, Hegab KH, El-Eraky W, El-Beih AA, Rehim MEA (2012) Synthesis of new transition metal complexes of 1H-perimidine derivatives having antimicrobial and anti-infammatory activities. Res ChemIntermed 38:1527–1550
CrossRef
- Farghaly, T.A., Abbas, E.M., Dawood, K.M. and El-Naggar, T., 2014. Synthesis of 2- phenylazonaphtho [1,8-ef][1,4] diazepines and 9-(3-arylhydrazono) pyrrolo [1,2-a] perimidines as antitumor agents. Molecules, 19(1), pp.740-755.
CrossRef
- Luthin, D.R., Rabinovich, A.K., Bhumralkar, D.R., Youngblood, K.L., Bychowski, R.A., Dhanoa, D.S. and May, J.M., 1999. Synthesis and biological activity of oxo-7H-benzo [e] perimidine-4-carboxylic acid derivatives as potent, nonpeptidecorticotropin releasing factor (CRF) receptor antagonists. Bioorganic & medicinal chemistry letters, 9(5), pp.765-770.
CrossRef
- Panchasara, D.R. and Pande, S., 2009. Synthesis and Biological Activity of 3-Chloro-1-(4-perimidine methylcarbonylamino)-4-phenyl-azetidin-2-one. E-Journal of Chemistry, 6(S1), pp.S91-S96.
CrossRef
- Alıcı B, Özdemir I, Karaaslan K, Çetinkaya E, Çetinkaya B (2005) Synthesis and catalytic properties of 1-alkylperimidineruthenium (II) complexes. J Mol CatalAChem 231(1–2):261–264 19.
CrossRef
- Choi G, Tsurugi H, Mashima K (2013) Hemilabile N-xylyl-N′-methylperimidinecarbene iridium complexes as catalysts for C–H activation and dehydrogenativesilylation: dual role of N-xylyl moiety for ortho-C–H bond activation and reductive bond cleavage. J Am ChemSoc 135(35):13149–13161
CrossRef
- Akıncı PA, Gülcemal S, Kazheva ON, Alexandrov GG, Dyachenko OA, Çetinkaya E, Çetinkaya B (2014) Perimidin-2-ylidene rhodium (I) complexes; unexpected halogen exchange and catalytic activities in transfer hydrogenation reaction. J OrganometalChem 765:23–30
CrossRef
- Jakubek M, Kejík Z, Kaplánek R, Veselá H, Sýkora D, Martásek P, Král V (2018) Perimidinebased synthetic receptors for determination of copper(II) in water solution. SupramolChem 30(3):218–226
CrossRef
- Malherbe RF (1983) 2,3-Dihydroperimidines as antioxidants for lubricants. US Patent
- Adole, V.A., Waghchaure, R.H., Pathade, S.S., Patil, M.R., Pawar, T.B. and Jagdale, B.S., 2020. Solvent-free grindstone synthesis of four new (E)-7-(arylidene)-indanones and their structural, spectroscopic and quantum chemical study: a comprehensive theoretical and experimental exploration. Molecular Simulation, 46(14), pp.1045-1054.
CrossRef
- Pathade, S.S. and Jagdale, B.S., 2020. Synthesis and DFT based quantum chemical studies of 2-(3-bromophenyl)-4-(4-bromophenyl)-2,3-dihydro-1H-1,5-benzodiazepine. J. Adv. Sci. Res, 11, pp.87-94.
- Adole, V.A., Pawar, T.B. and Jagdale, B.S., 2020. DFT computational insights into structural, electronic and spectroscopic parameters of 2-(2-Hydrazineyl) thiazole derivatives: a concise theoretical and experimental approach. Journal of Sulfur Chemistry, pp.1-18.
CrossRef
- Dhonnar, S.L., Adole, V.A., Sadgir, N.V. and Jagdale, B.S., 2021. Structural, Spectroscopic (UV-Vis and IR), Electronic and Chemical Reactivity Studies of (3,5-Diphenyl-4,5-dihydro-1H-pyrazol-1-yl)(phenyl)methanone. Physical Chemistry Research, 9(2), pp.193-209.
CrossRef
- Hiremath, S.M., Patil, A.S., Hiremath, C.S., Basangouda, M., Khemalapure, S.S., Patil, N.R., Radder, S.B., Armaković, S.J. and Armaković, S., 2019. Structural, spectroscopic characterization of 2-(5-methyl-1-benzofuran-3-yl) acetic acid in monomer, dimer and identification of specific reactive, drug likeness properties: experimental and computational study. Journal of Molecular Structure, 1178, pp.1-17.
- Abraham, C.S., Prasana, J.C., Muthu, S. and Raja, M., 2018. Quantum computational studies, spectroscopic (FT-IR, FT-Raman and UV–Vis) profiling, natural hybrid orbital and molecular docking analysis on 2,4Dibromoaniline. Journal of Molecular Structure, 1160, pp.393-405.
CrossRef
- Pandey, M., Muthu, S. and Gowda, N.N., 2017. Quantum mechanical and spectroscopic (FT-IR, FT-Raman, 1H, 13C NMR, UV-Vis) studies, NBO, NLO, HOMO, LUMO and Fukui function analysis of 5-Methoxy-1H-benzo[d]imidazole-2(3H)-thione by DFT studies. Journal of Molecular Structure, 1130, pp.511-521.
CrossRef
- Mofidi, F. and Reisi-Vanani, A., 2020. Investigation of the electronic and structural properties of graphyne oxide toward CO, CO2 and NH3 adsorption: A DFT and MD study. Applied Surface Science, 507, p.145134.
CrossRef
- Aghdasi, P., Ansari, R., Yousefi, S. and Goli, M., 2020. Structural and mechanical properties of pristine and adsorbed puckered arsenene nanostructures: A DFT study. Superlattices and Microstructures, 139, p.106414.
CrossRef
- Ibrahim, S.M. and Al-Hossainy, A.F., 2021. Synthesis, structural characterization, DFT, kinetics and mechanism of oxidation of bromothymol blue: application to textile industrial wastewater treatment. Chemical Papers, 75(1), pp.297-309.
CrossRef
- Karthikeyan, N., Prince, J.J., Ramalingam, S. and Periandy, S., 2015. Electronic [UV–Visible] and vibrational [FT-IR, FT-Raman] investigation and NMR–mass spectroscopic analysis of terephthalic acid using quantum Gaussian calculations. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 139, pp.229-242.
CrossRef
- Srivastava, A., Mishra, R., Kumar, S., Dev, K., Tandon, P. and Maurya, R., 2015. Molecular structure, spectral investigation (1H NMR, 13C NMR, UV–Visible, FT-IR, FT-Raman), NBO, intramolecular hydrogen bonding, chemical reactivity and first hyperpolarizability analysis of formononetin [7-hydroxy-3(4-methoxyphenyl) chromone]: A quantum chemical study. Journal of Molecular Structure, 1084, pp.55-73.
CrossRef
- Anand, S., Sundararajan, R.S., Ramachandraraja, C., Ramalingam, S. and Durga, R., 2015. Molecular vibrational investigation [FT-IR, FT-Raman, UV–Visible and NMR] on Bis (thiourea) Nickel chloride using HF and DFT calculations. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 138, pp.203-215.
CrossRef
- De Toledo, T.A., Pizani, P.S., Da Silva, L.E., Teixeira, A.M.R. and Freire, P.T.C., 2015. Spectroscopy studies on Schiff base N,N′-bis (salicylidene)-1,2-phenylenediamine by NMR, infrared, Raman and DFT calculations. Journal of Molecular Structure, 1097, pp.106-111.
CrossRef
- Karabacak, M., Kose, E., Sas, E.B., Kurt, M., Asiri, A.M. and Atac, A., 2015. DFT calculations and experimental FT-IR, FT-Raman, NMR, UV–Vis spectral studies of 3-fluorophenylboronic acid. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 136, pp.306-320.
CrossRef
- Sas, E.B., Kose, E., Kurt, M. and Karabacak, M., 2015. FT-IR, FT-Raman, NMR and UV–Vis spectra and DFT calculations of 5-bromo-2-ethoxyphenylboronic acid (monomer and dimer structures). Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 137, pp.1315-1333.
CrossRef
- Gökce, H., Öztürk, N., Ceylan, Ü., Alpaslan, Y.B. and Alpaslan, G., 2016. Thiol–thione tautomeric analysis, spectroscopic (FT-IR, Laser-Raman, NMR and UV–vis) properties and DFT computations of 5-(3-pyridyl)-4H-1,2,4-triazole-3-thiol molecule. SpectrochimicaActa Part A: Molecular and Biomolecular Spectroscopy, 163, pp.170-180.
CrossRef
- Suvitha, A., Periandy, S. and Gayathri, P., 2015. NBO, HOMO–LUMO, UV, NLO, NMR and vibrational analysis of veratrole using FT-IR, FT-Raman, FT-NMR spectra and HF–DFT computational methods. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 138, pp.357-369.
CrossRef
- Latorre, F., Kupfer, S., Bocklitz, T., Kinzel, D., Trautmann, S., Gräfe, S. and Deckert, V., 2016. Spatial resolution of tip-enhanced Raman spectroscopy–DFT assessment of the chemical effect. Nanoscale, 8(19), pp.10229-10239.
CrossRef
- Bonales, L.J., Colmenero, F., Cobos, J. and Timón, V., 2016. Spectroscopic Raman characterization of rutherfordine: a combined DFT and experimental study. Physical Chemistry Chemical Physics, 18(24), pp.16575-16584.
CrossRef
- Arjunan, V., Marchewka, M.K., Raj, A., Yang, H. and Mohan, S., 2015. Structural and vibrational spectral investigations of melaminium glutarate monohydrate by FTIR, FT-Raman and DFT methods. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 135, pp.540-550.
CrossRef
- Bilkan, M.T., Yurdakul, Ş., Demircioğlu, Z. and Büyükgüngör, O., 2016. Crystal structure, FT-IR, FT-Raman and DFT studies on a novel compound [C10H9N3] 4AgNO3. Journal of Organometallic Chemistry, 805, pp.108-116.
CrossRef
- Pathade, S.S., Adole, V.A., Jagdale, B.S. and Pawar, T.B., 2020. Molecular structure, electronic, chemical and spectroscopic (UV-visible and IR) studies of 5-(4-chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-pyrazole: combined DFT and experimental exploration. Material Science Research India, 17(specialissue2020), pp.27-40.
- Shinde, R.A., Adole, V.A., Jagdale, B.S. and Pawar, T.B., 2020. Experimental and Theoretical Studies on the Molecular Structure, FT-IR, NMR, HOMO, LUMO, MESP, and Reactivity Descriptors of (E)-1-(2,3-Dihydrobenzo [b][1,4] dioxin-6-yl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one. Material Science Research India, 17(specialissue2020), pp.54-72.
CrossRef
- Adole, V.A., Waghchaure, R.H., Jagdale, B.S., Pawar, T.B. and Pathade, S.S., Molecular tructure, frontier molecular orbital and spectroscopic examination on dihydropyrimidinones: a comparative computational approach.Journal of Advanced Scientific Research, 2020. 11 (2), pp.64-70.
- Sadgir, N.V., Dhonnar, S.L., Jagdale, B., Waghmare, B. and Sadgir, C., 2020. Synthesis, Spectroscopic Characterization, Quantum Chemical Study and Antimicrobial Study of (2E)-3-(2,6-Dichlorophenyl)-1-(4-Fluoro)-Prop-2-En-1-One. Material Science Research India, 17(3), pp.281-293.
CrossRef
- Adole, V.A., Jagdale, B.S., Pawar, T.B. and Desale, B.S., 2020. Molecular structure, frontier molecular orbitals, MESP and UV–visible spectroscopy studies of Ethyl-4-(3, 4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate: A theoretical and experimental appraisal. Material Science Research India, 17(specialissue2020), pp.13-36.
CrossRef
- Sathish, M., Rajasekaran, L., Shanthi, D., Kanagathara, N., Sarala, S. and Muthu, S., 2021. Spectroscopic (FT-IR, FT-Raman, UV-Vis) Molecular Structure, Electronic, Molecular docking, and thermodynamic investigations of indole-3-carboxylic acid by DFT method. Journal of Molecular Structure, p.130182.
CrossRef
- Sawant, A.B. and Nirwan, R.S., 2012. Synthesis, characterization and DFT studies of 6, 8-dichloro-2-(4-chlorophenyl)-4H-chromen-4-one.Indian Journal of Pure and Applied Physics, 50(5), pp.308-313
- Devi, K.S., Subramani, P., Parthiban, S. and Sundaraganesan, N., 2020. One-pot synthesis, spectroscopic characterizations, quantum chemical calculations, docking and cytotoxicity of 1-((dibenzylamino) methyl) pyrrolidine-2, 5-dione. Journal of Molecular Structure, 1203, p.127403.
CrossRef
- Sadgir, N.V., Dhonnar, S.L., Jagdale, B.S. and Sawant, A.B., 2020. Synthesis, spectroscopic characterization, XRD crystal structure, DFT and antimicrobial study of (2E)-3-(2, 6-dichlorophenyl)-1-(4-methoxyphenyl)-prop-2-en-1-one. SN Applied Sciences, 2(8), pp.1-12.
CrossRef
- Halim, S.A. and Ibrahim, M.A., 2021. Synthesis, DFT computational insights on structural, optical, photoelectrical characterizations and spectroscopic parameters of the novel (2E)-3-(4-methoxy-5-oxo-5H-furo [3, 2-g] chromen-6-yl) acrylonitrile (MOFCA). Journal of Molecular Structure, 1223, p.129316.
CrossRef
- Halim, S.A. and Ibrahim, M.A., 2017. Synthesis, DFT calculations, electronic structure, electronic absorption spectra, natural bond orbital (NBO) and nonlinear optical (NLO) analysis of the novel 5-methyl-8H-benzo [h] chromeno [2, 3-b][1, 6] naphthyridine-6 (5H), 8-dione (MBCND). Journal of Molecular Structure, 1130, pp.543-558.
CrossRef
- İnkaya, E., Günnaz, S., Özdemir, N., Dayan, O., Dinçer, M. and Çetinkaya, B., 2013. Synthesis, spectroscopic characterization, X-ray structure and DFT studies on 2, 6-bis (1-benzyl-1H-benzo [d] imidazol-2-yl) pyridine. SpectrochimicaActa Part A: Molecular and Biomolecular Spectroscopy, 103, pp.255-263.
CrossRef
- Yashwantrao, G., Jejurkar, V.P., Kshatriya, R. and Saha, S., 2019. Solvent-Free, Mechanochemically Scalable Synthesis of 2,3-Dihydroquinazolin-4(1H)-one Using Brønsted Acid Catalyst. ACS Sustainable Chemistry & Engineering, 7(15), pp.13551-13558.
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr et al. “Gaussian 03, revision C. 02; Gaussian, Inc.: Wallingford, CT, 2004.
CrossRef
- Becke, A.D., 1993. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys, 98(492), pp.5648-5652.
- Lee, C., Yang, W. and Parr, R.G., 1988. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. B, 37(2), pp.785-789.
CrossRef
- Krishnan, R.B.J.S., Binkley, J.S., Seeger, R. and Pople, J.A., 1980. Self‐consistent molecular orbital methods. XX. A basis set for correlated wave functions. The Journal of chemical physics, 72(1), pp.650-654.
CrossRef
- McLean, A.D. and Chandler, G.S., 1980. Contracted gaussian-basis sets for molecular calculations. 1. 2nd row atoms. Z-11–18. J ChemPhys 72: 5639–5648.
- Dennington, R.D.I.I., Keith, T. and Millam, J., 2007. GaussView, Version 4.1. 2. Semichem Inc., Shawnee Mission, KS.
CrossRef
- Sarojini, K., Krishnan, H., Kanakam, C.C. and Muthu, S., 2012. Synthesis, X-ray structural, characterization, NBO and HOMO–LUMO analysis using DFT study of 4-methyl-N-(naphthalene-1-yl) benzene sulfonamide. SpectrochimicaActa Part A: Molecular and Biomolecular Spectroscopy, 96, pp.657-667.
- Patel, U.H., Gandhi, S.A., Patel, B.D., Modh, R.D., Patel, R.H., Yadav, J. and Desai, K.R., 2013. Synthesis, characterizations, molecular structure and DFT studies of 4-benzylidene-2-(2-chloro-phenyl)-5-methyl-2, 4-dihydro-pyrazol-3-one.819-826IJPAP Vol.51(12) [December 2013]CrossRef
- Zhao, J., Song, P., Cui, Y., Liu, X., Sun, S., Hou, S. and Ma, F., 2014. Effects of hydrogen bond on 2-aminopyridine and its derivatives complexes in methanol solvent. SpectrochimicaActa Part A: Molecular and Biomolecular Spectroscopy, 131, pp.282-287.
CrossRef
- Adole, V.A., Jagdale, B.S., Pawar, T.B. and Sawant, A.B., 2020. Experimental and theoretical exploration on single crystal, structural, and quantum chemical parameters of (E)‐7‐(arylidene)‐1,2,6,7‐tetrahydro‐8H‐indeno [5,4‐b] furan‐8‐one derivatives: A comparative study. Journal of the Chinese Chemical Society, 67(10), pp.1763-1777.
CrossRef
- Koopmans, T., 1934. The classification of wave functions and eigen-values to the single electrons of an atom. Physica, 1, pp.104-113.

This work is licensed under a Creative Commons Attribution 4.0 International License.