Ultrasound Assisted Synthesis, Molecular Structure,UV-Visible Assignments, MEP and Mulliken Charges Study of (E)-3-(4-chlorophenyl)-1-(4-methoxyphenyl) prop-2-en-1-one: Experimental and DFT Correlational
Rohit S. Shinde
Department of Chemistry, Mahatma Gandhi Vidyamandir’s Arts, Science and Commerce College,(Affiliated to Savitribai Phule Pune University, Pune (MH), India) Manmad, Taluka-Nandgaon, District- Nashik, India-423104.
Corresponding Author E-mail: chemistry.rss@gmail.com
DOI : http://dx.doi.org/10.13005/msri/180110
Article Publishing History
Article Received on : 23-Mar-2020
Article Accepted on : 13-Apr-2021
Article Published : 13 Apr 2021
Plagiarism Check: Yes
Reviewed by: Dr. Piyush J. Patel
Second Review by: Dr. Nutan Sadgir
Final Approval by: Dr. Subhasis Roy
Article Metrics
ABSTRACT:
Present investigation deals with the synthesis and density functional theory study (DFT) of a chalcone derivative; (E)-3-(4-chlorophenyl)-1-(4-methoxyphenyl)prop-2-en-1-one (CPMPP). The synthesis of a CPMPP has been carried out by the reaction of 4-methoxyacetophenone and 4-chlorobenzalehyde in ethanol at 30 ℃ under ultrasound irradiation. The structure of a synthesized chalcone is affirmed on the basis of FT-IT, 1H NMR and 13C NMR. The geometry of a CPMPP is optimized by using the density functional theory method at the B3LYP/6-31G(d,p) basis set. The optimized geometrical parameters like bond length and bond angles have been computed. The absorption energies, oscillator strength, and electronic transitions have been derived at the TD-DFT method at the B3LYP/6-31G(d,p) level of theory for B3LYP/6-31G(d p) optimized geometries. The effect of polarity on the absorption energies is discussed by computing UV-visible results in dichloromethane (DCM). Since theoretically obtained wavenumbers are typically higher than experimental wavenumbers, computed wavenumbers were scaled with a scaling factor, and vibrational assignments were made by comparing experimental wavenumbers to scaled theoretical wavenumbers. Quantum chemical parameters have been determined and examined. Molecular electrostatic potential (MEP) surface plot analysis has been carried out at the same level of theory. Mulliken atomic charge study is also discussed in the present study.
KEYWORDS:
, B3LYP/6-311G (d,p); Chalcone; DFT; HOMO-LUMO
Copy the following to cite this article:
Shinde R. S. Ultrasound AssistedSynthesis, Molecular Structure,UV-Visible Assignments, MEP and Mulliken Charges Study of (E)-3-(4-chlorophenyl)-1-(4-methoxyphenyl)prop-2-en-1-one: Experimental and DFT Correlational. Mat. Sci. Res. India;18(1).
|
Copy the following to cite this URL:
Shinde R. S. Ultrasound AssistedSynthesis, Molecular Structure,UV-Visible Assignments, MEP and Mulliken Charges Study of (E)-3-(4-chlorophenyl)-1-(4-methoxyphenyl)prop-2-en-1-one: Experimental and DFT Correlational. Mat. Sci. Res. India;18(1). Available from: https://bit.ly/32adyiP
|
Introduction
Chalcone is the trivial name given to the α,β-unsaturated ketones,which are synthesized by condensing an aromatic aldehyde with an substituted acetophenone in the presence of a base. Chalcones are not only important precursors in the synthesis of many biologically active molecules, but they also make up a significant part of natural products1-5. Chalcones as well as their synthetic analogues show large number of medicinal properties [6-10]. They are crucial in the production of a wide variety of remedial compounds. They have shown remarkable curative efficacy in the treatment of a variety of diseases. Chalcone-based derivatives have gotten a lot of attention because of their simple structures and diverse pharmacological effects 6-10. The synthesis of these compounds has been reported using a variety of techniques and schemes.Aldol condensation and Claisen- Schmidt reaction continue to be the most widely used processes for synthesis of chalcones. In the presence of an aqueous alcoholic alkali, Claisen-Schmidt condensation occurs between equimolar amounts of a substituted acetophenone and substituted aldehydes. Chalcones and their heterocyclic analogues have a wide range of pharmacological properties, including anticancer, antioxidant, anti-inflammatory, antifungal, antiviral, antibacterial, antiproliferative, antitumor, antimalarial, antidiabetic, anticonvulsant, and many others, making them a significant scaffold in medicinal chemistry 16-26. Green Chemistry has played vital role for designing many synthetic molecules without causing environmental hazards 27-37. In that ultrasound technique is proved to be highly efficient. DFT is a method that can provide a good deal of information regarding the physical and chemical behavior of the molecules 38-53. In this unique circumstance, I would like to present current research on ultrasound assisted synthesis, molecular structure, UV-visible assignments, MEP, and Mulliken charges study of (E)-3-(4-chlorophenyl)-1-(4-methoxyphenyl)prop-2-en-1-one: a DFT and experimental study correlational.
Experimental
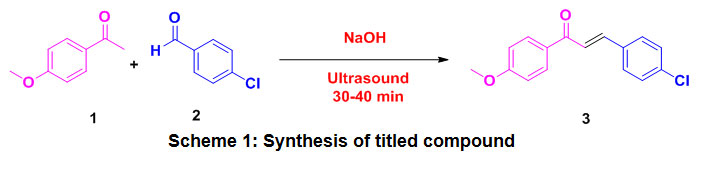
Chalcones were synthesized by base catalyzed Claisen‐Schmidt condensation reaction of appropriately 4-methoxyacetophenone1 and 4-chlorobenzalehyde2 by literature method. Anequimolar mixture of 4-methoxyacetophenone and 4-chlorobenzalehyde in 10 mL ethanol in a 50 mL conical flask equipped. Then appropriate amount of KOH solution was added drop wise to the reaction mixture. The alkaline mixture was exposed to ultrasound irradiation until formation of the product (checked by TLC). After this, reaction mixture was neutralized by 1:1 HCl whereby the precipitation occurred. On filtering off, the crude chalconewas dried in air and recrystallized by ethanol to give pale yellow crystals of chalcone 3.
Scheme 1: Synthesis of titled compound
Table 1: Physicochemical and Spectral data of CPMPP molecule
|
Systematic Name of the Product
|
(E)-3-(4-Chlorophenyl)-1-(4-methoxyphenyl)prop-2-en-1-one
|
|
Abbreviation used
|
CPMPP
|
|
Physicochemical data
|
Yield: 88%, Colour: pale yellow crystals,
|
|
FT-IR
IR (KBr, cm-1)
|
470, 532.35, 694.37, 771.53, 833, 995, 1087, 1211, 1327, 1489, 1597, 1666, 3047
|
|
1H NMR
(400 MHz, CDCl3)
|
3.90 (s, 3H), 6.97−7.01(m, 2H), 7.37−7.41 (m, 2H), 7.52 (d, J = 15.7 Hz, 1H), 7.55−7.59 (m, 2H), 7.75 (d, J = 15.7 Hz, 1H), 8.02−8.06 (m, 2H)
|
|
13C NMR
(100 MHz, CDCl3)
|
55.65, 114.05, 122.44, 129.33, 129.66, 130.98, 131.07, 133.74, 136.30, 142.55, 163.70, 188.48.
|
Computational study
DFT calculations were performed on an Intel (R) Core (TM) i5 computer using Gaussian-03 program package without any constraint on the geometry. The geometry of the molecules studied in this is optimized by DFT/B3LYP method using 6-31G(d,p) basis set [54]. The FMO analysis and quantum chemical study has been performed using same basis set. Absorption energies (λ in nm), Oscillator strength (ƒ), and Transitions of title molecule have been calculated at TD-B3LYP/6-31G(d,p) level of theory for B3LYP/6-31G(d,p) optimized geometries. To investigate the reactive sites of the title molecules, the molecular electrostatic potential (MEP) was computed using the same method. All the calculations were carried out for the optimized structure in the gas phase.
Results and Discussion
Optimized Molecular Structure
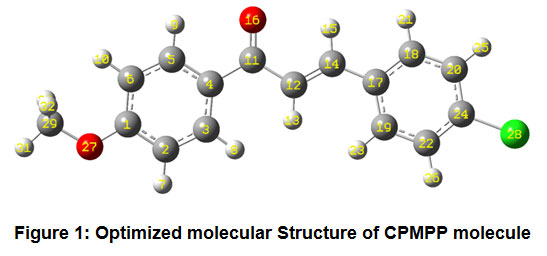
The optimized molecular structure of the title molecule CPMPP is given in Figure 1. The molecule CPMPP is having C1 point group symmetry and the dipole moment is 3.1533 Debye. The optimized geometrical parameters; bond lengths and bond angles of the title molecule have been computed and presented here in Table 2 and Table 3. In the molecule CPMPP, the C=O (C11-O16) bond length is 1.2314Å. The C-Cl (C24-Cl28) bond length is 1.7559 Å. The bond lengths in C-O are 1.359Å (C1-O27) and 1.422Å (O27-C29). Amongst aromatic C=C bond lengths, C17-C19 bond is the longest (1.4086Å) and the shortest is C2-C3 (1.3865Å). Other bond lengths are also in good agreement. All the bond angles are also in good agreement.
Figure 1: Optimized molecular Structure of CPMPP molecule
Table 2: Optimized Bond Lengths of CPMPPmolecule obtained at B3LYP/6-31G(d.p)
|
Bond lengths (Å)
|
|
C1-C2
|
1.4038
|
C14-C17
|
1.4615
|
|
C1-C6
|
1.4033
|
C17-C18
|
1.4068
|
|
C1-O27
|
1.359
|
C17-C19
|
1.4086
|
|
C2-C3
|
1.3865
|
C18-C20
|
1.392
|
|
C2-H7
|
1.0847
|
C18-H21
|
1.0863
|
|
C3-C4
|
1.4077
|
C19-C22
|
1.3892
|
|
C3- H8
|
1.0843
|
C19-H23
|
1.0851
|
|
C4- C5
|
1.4028
|
C20-C24
|
1.3935
|
|
C4-C11
|
1.4941
|
C20-H25
|
1.0841
|
|
C5- C6
|
1.3902
|
C22-C24
|
1.3972
|
|
C5- H9
|
1.0846
|
C22-H26
|
1.0842
|
|
C6- H10
|
1.0833
|
C24-Cl28
|
1.7559
|
|
C11-C12
|
1.4871
|
O27-C29
|
1.422
|
|
C11-O16
|
1.2314
|
C29-H30
|
1.0968
|
|
C12-H13
C12-C14
C14-H15
|
1.084
1.3472
1.0889
|
C29-H31
C29-H32
–
|
1.0905
1.0968
–
|
Table 3: Optimized Bond Angles of CPMPPmolecule obtained at B3LYP/6-31(d,p)
|
Bond Angles (o)
|
|
C2-C1-C6
|
119.6544
|
H15-C14-C17
|
116.1539
|
|
C2-C1-O27
|
115.6855
|
C14-C17-C18
|
118.6637
|
|
C6-C1-O27
|
124.66
|
C14-C17-C19
|
123.5224
|
|
C1-C2-C3
|
120.158
|
C18-C17-C19
|
117.8139
|
|
C1-C2-H7
|
118.4593
|
C17-C18-C20
|
121.6314
|
|
C3-C2-H7
|
121.3826
|
C17-C18-H21
|
119.1249
|
|
C2-C3-C4
|
121.0568
|
C20-C18-H21
|
119.2437
|
|
C2-C3-H8
|
118.1341
|
C17-C19-C22
|
121.2893
|
|
C4-C3-H8
|
120.8024
|
C17-C19-H23
|
120.011
|
|
C3-C4-C5
|
117.9523
|
C22-C19-H23
|
118.6996
|
|
C3-C4-C11
|
124.4406
|
C18-C20-C24
|
118.9658
|
|
C5-C4-C11
|
117.6033
|
C18-C20-H25
|
120.8546
|
|
C4-C5-C6
|
121.7417
|
C24-C20-H25
|
120.1795
|
|
C4-C5-H9
|
117.7734
|
C19-C22-C24
|
119.3094
|
|
C6-C5-H9
|
120.4849
|
C19-C22-H26
|
120.7342
|
|
C1-C6-C5
|
119.4349
|
C24-C22-H26
|
119.9564
|
|
C1-C6-H10
|
120.9931
|
C20-C24-C22
|
120.9901
|
|
C5-C6-H10
|
119.572
|
C20-C24-Cl28
|
119.6102
|
|
C4-C11-C12
|
119.3039
|
C22-C24-Cl28
|
119.3997
|
|
C4-C11-O16
|
120.1517
|
C1-O27-C29
|
118.5306
|
|
C12-C11-O16
|
120.5424
|
O27-C29-H30
|
111.5468
|
|
C11-C12-H13
|
119.0571
|
O27-C29-H31
|
105.8923
|
|
C11-C12-C14
|
119.8758
|
O27-C29-H32
|
111.551
|
|
H13-C12-C14
|
121.0615
|
H30-C29-H31
|
109.2772
|
|
C12-C14-H15
|
115.8011
|
H30-C29-H32
|
109.2239
|
|
C12-C14-C17
|
128.045
|
–
|
–
|
Global descriptors study
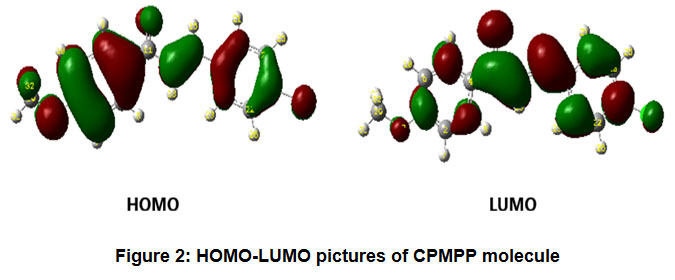
The pictorial representation of HOMO-LUMO orbitals is given in Figure 2. The electronic parameters such as EHOMO, ELUMO, ionization enthalpy (I), and electron affinity (A) are given in Table 4. The quantum chemical parameters likeelectronegativity (χ), absolute hardness (η), softness (σ), electrophilicity (ω), chemical potential (Pi) are presented in Table 5. The frontier molecular orbital (FMO) analysis suggests that the energy gap in the molecule CPMPP is 4.06 eV. The lower HOMO-LUMO energy gap demonstrates the inevitable charge transfer is happening within the molecule. The global softness (σ), and the absolute hardness (η) values are 0.4926and 2.03 eV respectively. The ease of removal of an electron is governed by its chemical potential Pi and it is likewise identified with its electronegativity (χ). A good electrophile is described by a higher value of global electrophilicity (ω) and the higher value of ω indicates good nucleophile. Our results suggest that the molecule CPMPP has a higher value of global electrophilicity (ω = 4.2625eV), so it is most likely to accept electrons readily and also would undergo nucleophilic attack easily. As Pi value increases, the ability of a molecule to lose an electron increases. The maximum charge transfer is in the title molecule is 2.0493eV.
Figure 2: HOMO-LUMO pictures of CPMPP molecule
Table 4: Electronic parameters of CPMPP molecule
|
Entry
|
E
(a.u.)
|
EHOMO
(eV)
|
ELUMO
(eV)
|
I
(eV)
|
A
(eV)
|
Eg
(eV)
|
|
CPMPP
|
-1228.18
|
-6.19
|
-2.13
|
6.19
|
2.13
|
4.06
|
Table 5: Global reactivity parameters of CPMP Pmolecule.
|
Entry
|
χ
(eV)
|
ɳ
(eV)
|
σ
(eV-1)
|
ω
(eV)
|
Pi
(eV)
|
ΔNmax
(eV)
|
Dipole Moment (Debye)
|
|
CPMPP
|
4.16
|
2.03
|
0.4926
|
4.2625
|
-4.16
|
2.0493
|
3.1533
|
UV-Visible Analysis
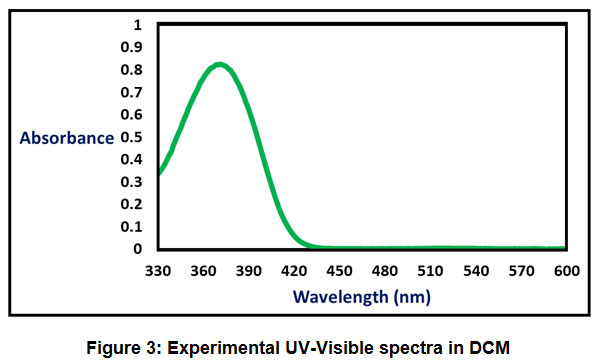
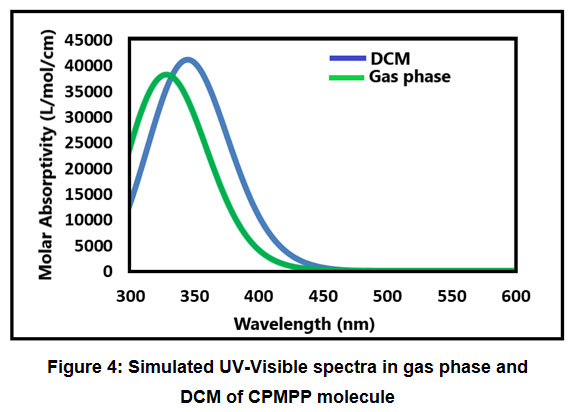
The absorption energies (λ in nm), oscillator strength (ƒ) and electronic transitions of the CPMP Pmolecule were computed at the TD-DFT-B3LYP/6-31G(d,p) level of theory for the optimized structure. The absorption energies (λ in nm), oscillator strength (ƒ) and transitions of CPMP Pmolecule and experimental UV-Vis spectral analysis are given in Table 6. The theoretical UV-Vis absorption results were simulated up to three singlet excited states. The experimental UV-Vis spectra were recorded in the DCM solvent. Likewise, the theoretical UV-Vis spectra were computed in the gas phase, and the DCM solvent. The theoretical and experimental UV-Vis spectral images are depicted in Figure 3 and Figure4. The first singlet state (S1) is found to be at 382.50 nm in the gas phase and 368.76 nm in DCM. The experimental UV-visible absorption band is centred at 375.02nm in DCM solvent. This result suggests that the theoretical UV-Vis absorption results are in acceptable concurrence with the UV-Vis absorption experimental results. The HOMO-LUMO electronic transition corresponds to the 71 -> 72 configurations. The solvent effect on the HOMO-LUMO absorption wavelength of the DPPPM molecule is found to be a blue shift (hypsochromic shift) as per the UV-Vis absorption theoretical data. The second singlet excited state (S2) is present at 338.93nm (gas phase) and 350.05 nm (DCM) in the theoretical UV-Vis absorption spectrum. The second gas phase singlet excited state and also DCMO is composed of only oneconfiguration, namely 71 -> 72 configurations. The third gas phase singlet excited state (S3) is from 69 -> 72, 70 -> 72, and 71 -> 73 and 69 -> 72, 70 -> 72 in DCM. The third absorption band is located at 307.68 nm (gas phase) and 315.08 nm (DCM).
Figure 3: Experimental UV-Visible spectra in DCM
Figure 4: Simulated UV-Visible spectra in gas phase and DCM of CPMPP molecule
Table 6 :Absorption wavelength (λ in nm), coefficient, oscillator strength (ƒ), and electronic transitions of titled compound computed at TD-DFT B3LYP/6-31G(d,p) level of theory (Experimental value of absorption wavelength (λ in nm) given in bracket).
|
Gas Phase
|
DCM
|
|
State
|
Config
|
Coefficient
|
f
|
λ, nm
|
Config
|
Coefficient
|
f
|
λ, nm
|
|
I
|
69 -> 72 69 -> 73 70 -> 72
|
0.67881
0.10098
-0.12390
|
0.0002
|
382.50
|
69 -> 72
70 -> 72
|
0.68263
0.10238
|
0.0019
|
368.76
(375.02)
|
|
II
|
71 -> 72
|
0.70176
|
0.6207
|
338.93
|
71 -> 72
|
0.70087
|
0.8907
|
350.05
|
|
III
|
69 -> 72 70 -> 72
71 -> 73
|
0.12029
0.67825
-0.10067
|
0.4056
|
307.68
|
69 -> 72 70 -> 72
|
-0.10306
0.68537
|
0.2899
|
315.08
|
Config- Configuration
Vibrational study
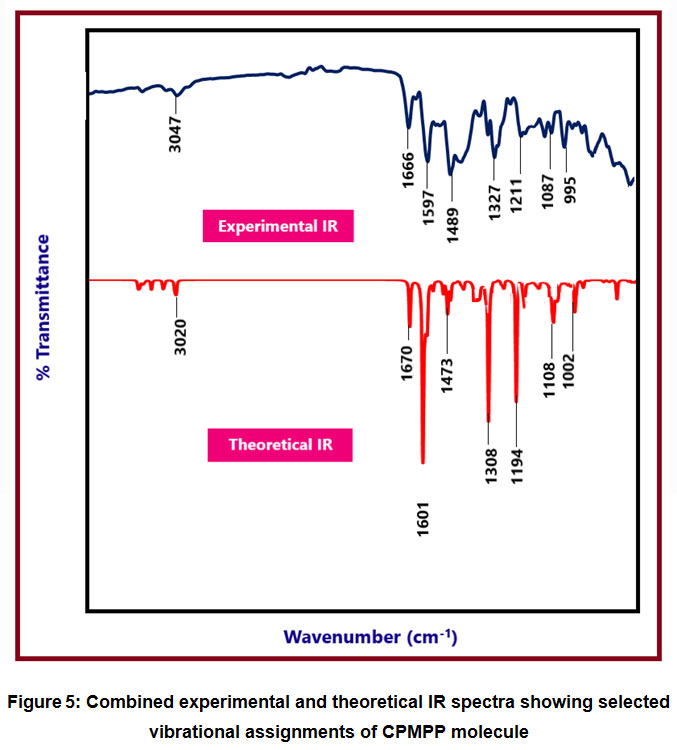
The experimental and theoretical IR spectrum of the title compound is given in Figure 5. In the given spectra, scaled theoretical IR values and experimental IR values are depicted. The comparison between the experimental and scaled IR values has led to the correct assignment of the vibrational bands. The CPMPP molecule consists of 32 atoms with 90 normal modes of vibration. Because of the anharmonicity of the incomplete treatment of electron correlation and the use of a finite one-particle basis set, computed harmonic vibrational wavenumbers are usually higher than experimental ones 55. B3LYP method was used to calculate harmonic frequencies using 6-31G(d,p) basis sets, which were then scaled by appropriate scaling factor 56. The BLYP/6-31G(d,p) method produced better results, with a deviation from the experiment of less than 30 cm-1.
The aromatic C-H vibrational stretching is located at 3047 cm-1 in the in the experimental IR spectrum and at 3020 cm-1 theoretically predicted IR spectrum. This definitely suggests a great deal of agreement between the two compared values. The C=C stretching vibrations is observed at 1601 cm-1 and 1597 cm-1 in the experimental and theoretical IR spectra respectively. The another important C=C vibration was found at 1489 cm-1 and 1473 cm-1 in the experimental and theoretical IR spectra respectively. The C-H bending vibrations are found at 1327 cm-1 and 1308 cm-1 in the experimental and theoretical IR spectra respectively. The C-O stretching vibration is found at 1211 cm-1 in the experimental IR spectrum and at1194 cm-1 in the theoretical IR spectrum. The 1087 cm-1 in the experimental and 1108 in the experimental are attributed to C-C stretching vibrations. The experimental 995 cm-1 matches with the theoretical 1002 cm-1 is occurred due to the bending vibrational mode.Throughout this regard, the correlation was used to correctly assign vibrational bands to the title compounds, and the IR spectra of the titled compound have a very strong correlation.
Figure 5: Combined experimental and theoretical IR spectra showing selected vibrational assignments of CPMPP molecule
Mulliken Atomic Charges and Molecular Electrostatic Plot Analysis
The Mulliken atomic charges of the CPMPP molecule are calculated by DFT/B3LYP method with 6-31G(d,p) basis set in the gaseous phase and are given in Table 7. Mulliken atomic charges reveal that all the hydrogen atoms have a net positive charge but H15 and H31 atoms have a more positive charge than other hydrogen atoms and therefore they are more acidic. Amongst, carbon atoms, the C11 atom have the highest net positive charge (0.372919) as it is attached to an electronegative oxygen atom. On the other hand C12 atom has the highest negative charge (-0.141075).
Table 7: Mulliken atomic chargesof CPMPP
|
Atom
|
Charge
|
Atom
|
Charge
|
|
1 C
|
0.362574
|
17 C
|
0.127545
|
|
2 C
|
-0.128205
|
18 C
|
-0.123597
|
|
3 C
|
-0.112476
|
19 C
|
-0.105209
|
|
4 C
|
0.038662
|
20 C
|
-0.073641
|
|
5 C
|
-0.107676
|
21 H
|
0.104888
|
|
6 C
|
-0.137000
|
22 C
|
-0.074632
|
|
7 H
|
0.098792
|
23 H
|
0.094380
|
|
8 H
|
0.087747
|
24 C
|
-0.093211
|
|
9 H
|
0.120507
|
25 H
|
0.115548
|
|
10 H
|
0.092396
|
26 H
|
0.114129
|
|
11 C
|
0.372919
|
27 O
|
-0.512859
|
|
12 C
|
-0.141075
|
28 Cl
|
-0.013042
|
|
13 H
|
0.081235
|
29 C
|
-0.083420
|
|
14 C
|
-0.090049
|
30 H
|
0.118400
|
|
15 H
|
0.122194
|
31 H
|
0.129564
|
|
16 O
|
-0.503478
|
32 H
|
0.118091
|
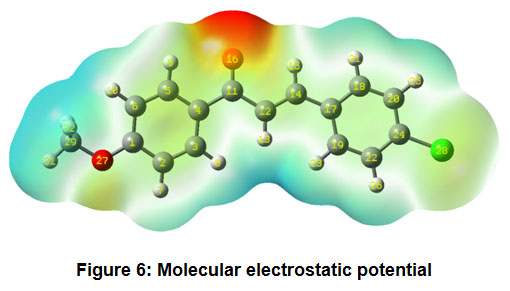
Figure 6 shows the molecular electrostatic potential plot. The use of a molecular electrostatic potential can be used to evaluate phenomena such as nucleophilic and electrophilic positions, solvent effects, hydrogen bonding interactions, and so on. MEP is mainly used to establish the reactive sites of molecules, enabling researchers to predict how one particle will interact with another. The various electrostatic potential values at the molecule’s surface are expressed by different colours. Electrophilic reactivity is correlated with the red and yellow zones, which lead to high electron density. The blue sections, on the other hand, reflect low electron density and nucleophilic reactivity, while the green colours represent regions of zero potential.
Figure 6: Molecular electrostatic potential
Conclusion
Present investigation deals with the synthesis and DFT study of a chalcone derivative; (E)-3-(4-chlorophenyl)-1-(4-methoxyphenyl)prop-2-en-1-one. The synthesis of a CPMPP has been carried out by the reaction of 4-methoxyacetophenone and 4-chlorobenzalehyde in ethanol at 30℃ under ultrasound irradiation. The structure of a synthesized chalcone is affirmed on the basis of FT-IT, 1H NMR and 13C NMR. The geometry of a CPMPP is optimized by using the density functional theory method at the B3LYP/6-31G(d,p) basis set. The molecule CPMPP is having C1 point group symmetry and the dipole moment is 3.1533 Debye. The optimized geometrical parameters like bond length and bond angles have been computed. The quantum chemical parameters likeelectronegativity, absolute hardness, softness, electrophilicity, chemical potential are presented. The FMO analysis suggests that the energy gap in the molecule CPMPP is 4.06 eV. The lower HOMO-LUMO energy gap demonstrates the inevitable charge transfer is happening within the molecule.The absorption energies, oscillator strength, and electronic transitions have been derived at the TD-DFT method at the B3LYP/6-31G(d,p) level of theory for B3LYP/6-31G(d p) optimized geometries. The effect of polarity on the absorption energies is discussed by computing UV-visible results in DCM. The solvent effect on the HOMO-LUMO absorption wavelength of the CPMPP molecule is found to be a blue shift as per the UV-Vis absorption theoretical data. Mulliken atomic charges reveal that all the hydrogen atoms have a net positive charge but H15 and H31 atoms have a more positive charge than other hydrogen atoms and therefore they are more acidic. Amongst, carbon atoms, the C11 atom have the highest net positive charge (0.372919) as it is attached to an electronegative oxygen atom. On the other hand C12 atom has the highest negative charge (-0.141075).
Acknowledgment
Author acknowledges Department of Chemistry, Arts, Science and Commerce College, Manmad, MS, India for the research assistance. Authoris grateful to Prof. (Dr.) T. B. Pawar for his guidance for the Gaussian study.
Funding Sourse
No funding was received to carry out the present research.
References
- MT Albuquerque, H., MM Santos, C., AS Cavaleiro, J. and MS Silva, A., 2014. Chalcones as Versatile Synthons for the Synthesis of 5-and 6-membered Nitrogen Heterocycles. Current Organic Chemistry, 18(21), pp.2750-2775.
CrossRef
- Velikorodov, A.V., Ionova, V.A., Temirbulatova, S.I., Titova, O.L. and Stepkina, N.N., 2013. Synthesis and application of chalcones to the preparation of heterocyclic structures. Russian journal of organic chemistry, 49(11), pp.1610-1616.
CrossRef
- Rostom, S.A., Badr, M.H., Abd El Razik, H.A., Ashour, H.M. and Abdel Wahab, A.E., 2011. Synthesis of some pyrazolines and pyrimidines derived from polymethoxy chalcones as anticancer and antimicrobial agents. Archiv der Pharmazie, 344(9), pp.572-587.
CrossRef
- Adel, A.H., Ahmed, E.S., Hawata, M.A., Kasem, E.R. and Shabaan, M.T., 2007. Synthesis and Antimicrobial Evaluation of Some Chalcones and Their Derived Pyrazoles, Pyrazolines, Isoxazolines, and 5,6-Dihydropyrimidine-2-(1H)-thiones. Monatshefte für Chemie-Chemical Monthly, 138(9), pp.889-897.
CrossRef
- Chobe, S.S., Adole, V.A., Deshmukh, K.P., Pawar, T.B. and Jagdale, B.S., 2014. Poly (ethylene glycol)(PEG-400): A green approach towards synthesis of novel pyrazolo [3, 4-d] pyrimidin-6-amines derivatives and their antimicrobial screening. Archives of Applied Science Research, 6(2), pp.61-66.
- Cui, M., Ono, M., Kimura, H., Liu, B.L. and Saji, H., 2011. Synthesis and biological evaluation of indole-chalcone derivatives as β-amyloid imaging probe. Bioorganic & medicinal chemistry letters, 21(3), pp.980-982.
CrossRef
- Sun, L.P., Jiang, Z., Gao, L.X., Sheng, L., Quan, Y.C., Li, J. and Piao, H.R., 2013. Synthesis and biological evaluation of furan-chalcone derivatives as protein tyrosine phosphatase inhibitors. Bulletin of the Korean Chemical Society, 34(4), pp.1023-1024.
CrossRef
- Dandawate, P., Ahmed, K., Padhye, S., Ahmad, A. and Biersack, B., 2021. Anticancer Active heterocyclic chalcones: recent developments. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents), 21(5), pp.558-566.
CrossRef
- Kuete, V., Nkuete, A.H., Mbaveng, A.T., Wiench, B., Wabo, H.K., Tane, P. and Efferth, T., 2014. Cytotoxicity and modes of action of 4′-hydroxy-2′, 6′-dimethoxychalcone and other flavonoids toward drug-sensitive and multidrug-resistant cancer cell lines. Phytomedicine, 21(12), pp.1651-1657.
CrossRef
- Adole, V.A., Pawar, T.B. and Jagdale, B.S., 2020. Aqua‐mediated rapid and benign synthesis of 1,2,6,7‐tetrahydro‐8H‐indeno [5,4‐b] furan‐8‐one‐appended novel 2‐arylidene indanones of pharmacological interest at ambient temperature . Journal of the Chinese Chemical Society, 67(2), pp.306-315.
CrossRef
- Chiaradia, L.D., Dos Santos, R., Vitor, C.E., Vieira, A.A., Leal, P.C., Nunes, R.J., Calixto, J.B. and Yunes, R.A., 2008. Synthesis and pharmacological activity of chalcones derived from 2, 4, 6-trimethoxyacetophenone in RAW 264.7 cells stimulated by LPS: Quantitative structure–activity relationships. Bioorganic & medicinal chemistry, 16(2), pp.658-667.
CrossRef
- Batovska, D.I. and Todorova, I.T., 2010. Trends in utilization of the pharmacological potential of chalcones. Current clinical pharmacology, 5(1), pp.1-29.
CrossRef
- Nowakowska, Z., 2007. A review of anti-infective and anti-inflammatory chalcones. European journal of medicinal chemistry, 42(2), pp.125-137.
CrossRef
- K Sahu, N., S Balbhadra, S., Choudhary, J. and V Kohli, D., 2012. Exploring pharmacological significance of chalcone scaffold: a review. Current medicinal chemistry, 19(2), pp.209-225.
CrossRef
- Matos, M.J., Vazquez-Rodriguez, S., Uriarte, E. and Santana, L., 2015. Potential pharmacological uses of chalcones: a patent review (from June 2011–2014). Expert opinion on therapeutic patents, 25(3), pp.351-366.
CrossRef
- Syam, S., Abdelwahab, S.I., Al-Mamary, M.A. and Mohan, S., 2012. Synthesis of chalcones with anticancer activities . Molecules, 17(6), pp.6179-6195.
CrossRef
- Anto, R.J., Sukumaran, K., Kuttan, G., Rao, M.N.A., Subbaraju, V. and Kuttan, R., 1995. Anticancer and antioxidant activity of synthetic chalcones and related compounds. Cancer letters, 97(1), pp.33-37.
CrossRef
- Won, S.J., Liu, C.T., Tsao, L.T., Weng, J.R., Ko, H.H., Wang, J.P. and Lin, C.N., 2005. Synthetic chalcones as potential anti-inflammatory and cancer chemopreventive agents. European journal of medicinal chemistry, 40(1), pp.103-112.
CrossRef
- Lahtchev, K.L., Batovska, D.I., St P, P., Ubiyvovk, V.M. and Sibirny, A.A., 2008. Antifungal activity of chalcones: A mechanistic study using various yeast strains. European journal of medicinal chemistry, 43(10), pp.2220-2228.
CrossRef
- Trivedi, J.C., Bariwal, J.B., Upadhyay, K.D., Naliapara, Y.T., Joshi, S.K., Pannecouque, C.C., De Clercq, E. and Shah, A.K., 2007. Improved and rapid synthesis of new coumarinyl chalcone derivatives and their antiviral activity. Tetrahedron Letters, 48(48), pp.8472-8474.
CrossRef
- Ávila, H.P., Smânia, E.D.F.A., Delle Monache, F. and Júnior, A.S., 2008. Structure–activity relationship of antibacterial chalcones. Bioorganic & medicinal chemistry, 16(22), pp.9790-9794.
CrossRef
- Boumendjel, A., Boccard, J., Carrupt, P.A., Nicolle, E., Blanc, M., Geze, A., Choisnard, L., Wouessidjewe, D., Matera, E.L. and Dumontet, C., 2008. Antimitotic and antiproliferative activities of chalcones: forward structure–activity relationship. Journal of medicinal chemistry, 51(7), pp.2307-2310.
CrossRef
- Kumar, D., Kumar, N.M., Akamatsu, K., Kusaka, E., Harada, H. and Ito, T., 2010. Synthesis and biological evaluation of indolyl chalcones as antitumor agents. Bioorganic & medicinal chemistry letters, 20(13), pp.3916-3919.
CrossRef
- Li, R., Kenyon, G.L., Cohen, F.E., Chen, X., Gong, B., Dominguez, J.N., Davidson, E., Kurzban, G., Miller, R.E., Nuzum, E.O. and Rosenthal, P.J., 1995. In vitro antimalarial activity of chalcones and their derivatives. Journal of medicinal chemistry, 38(26), pp.5031-5037.
CrossRef
- Rammohan, A., Bhaskar, B.V., Venkateswarlu, N., Gu, W. and Zyryanov, G.V., 2020. Design, synthesis, docking and biological evaluation of chalcones as promising antidiabetic agents. Bioorganic chemistry, 95, p.103527.
CrossRef
- Parmar, S.S., Pandey, B.R., Dwivedi, C. and Harbison, R.D., 1974. Anticonvulsant activity and monoamine oxidase inhibitory properties of 1, 3, 5‐trisubstituted pyrazolines. Journal of pharmaceutical sciences, 63(7), pp.1152-1155.
CrossRef
- El-Borai, M.A., Rizk, H.F., Sadek, M.R. and El-Keiy, M.M., 2016. An eco-friendly synthesis of heterocyclic moieties condensed with pyrazole system under green conditions and their biological activity. Green and Sustainable Chemistry, 6(2), pp.88-100.
CrossRef
- Shinde, R.A., Adole, V.A., Jagdale, B.S. and Pawar, T.B., 2020. Experimental and Theoretical Studies on the Molecular Structure, FT-IR, NMR, HOMO, LUMO, MESP, and Reactivity Descriptors of (E)-1-(2,3-Dihydrobenzo [b][1,4] dioxin-6-yl)-3-(3,4,5-trimethoxyphenyl) prop-2-en-1-one. Material Science Research India, 17(specialissue2020), pp.54-72.
CrossRef
- Abbass, E.M., Khalil, A.K. and El‐Naggar, A.M., 2020. Eco‐friendly synthesis of novel pyrimidine derivatives as potential anticancer agents. Journal of Heterocyclic Chemistry, 57(3), pp.1154-1164.
CrossRef
- Adole, V.A., Waghchaure, R.H., Pathade, S.S., Patil, M.R., Pawar, T.B. and Jagdale, B.S., 2020. Solvent-free grindstone synthesis of four new (E)-7-(arylidene)-indanones and their structural, spectroscopic and quantum chemical study: a comprehensive theoretical and experimental exploration. Molecular Simulation, 46(14), pp.1045-1054.
CrossRef
- Rayudu, S.V., Karmakar, D. and Kumar, P., 2019. Water-acetic acid mediated an efficient one-pot eco-friendly synthesis of novel bis-isoxazolopyrroloquinoline derivatives. Tetrahedron Letters, 60(36), p.151025.
CrossRef
- Shinde, R.A., Adole, V.A., Jagdale, B.S., Pawar, T.B., Desale, B.S. and Shinde, R.S., 2020. Efficient Synthesis, Spectroscopic and Quantum Chemical Study of 2, 3-Dihydrobenzofuran Labelled Two Novel Arylidene Indanones: A Comparative Theoretical Exploration. Material Science Research India, 17(2), pp.146-161.
CrossRef
- Pourshojaei, Y., Jadidi, M.H., Eskandari, K., Foroumadi, A. and Asadipour, A., 2018. An eco-friendly synthesis of 4-aryl-substituted pyrano-fuzed coumarins as potential pharmacological active heterocycles using molybdenum oxide nanoparticles as an effective and recyclable catalyst. Research on Chemical Intermediates, 44(7), pp.4195-4212.
CrossRef
- Adole, V.A., Pawar, T.B., Koli, P.B. and Jagdale, B.S., 2019. Exploration of catalytic performance of nano-La2O3 as an efficient catalyst for dihydropyrimidinone/thione synthesis and gas sensing. Journal of Nanostructure in Chemistry, 9(1), pp.61-76.
CrossRef
- Adole, V.A., More, R.A., Jagdale, B.S., Pawar, T.B. and Chobe, S.S., 2020. Efficient synthesis, antibacterial, antifungal, antioxidant and cytotoxicity study of 2‐(2‐hydrazineyl) thiazole derivatives. ChemistrySelect, 5(9), pp.2778-2786.
CrossRef
- Uludağ, N. and Serdaroğlu, G., 2018. An improved synthesis, spectroscopic (FT-IR, NMR) study and DFT computational analysis (IR, NMR, UV–Vis, MEP diagrams, NBO, NLO, FMO) of the 1, 5-methanoazocino [4, 3-b] indole core structure. Journal of Molecular Structure, 1155, pp.548-560.
CrossRef
- Adole, V.A., 2020. Synthetic approaches for the synthesis of dihydropyrimidinones/thiones (biginelli adducts): a concise review. World Journal of Pharmaceutical Research, 9(6), pp.1067-1091.
- Kaviani, S., Izadyar, M. and Housaindokht, M.R., 2016. Solvent and spin state effects on molecular structure, IR spectra, binding energies and quantum chemical reactivity indices of deferiprone–ferric complex: DFT study. Polyhedron, 117, pp.623-627.
CrossRef
- Dhonnar, S.L., Adole, V.A., Sadgir, N.V. and Jagdale, B.S., 2021. Structural, Spectroscopic (UV-Vis and IR), Electronic and Chemical Reactivity Studies of (3, 5-Diphenyl-4, 5-dihydro-1H-pyrazol-1-yl)(phenyl) methanone. Physical Chemistry Research, 9(2), pp.193-209.
CrossRef
- Sheikhi, M., Shahab, S., Khaleghian, M., Hajikolaee, F.H., Balakhanava, I. and Alnajjar, R., 2018. Adsorption properties of the molecule resveratrol on CNT (8, 0-10) nanotube: geometry optimization, molecular structure, spectroscopic (NMR, UV/Vis, excited state), FMO, MEP and HOMO-LUMO investigations. Journal of Molecular Structure, 1160, pp.479-487.
CrossRef
- Adole, V.A., Waghchaure, R.H., Jagdale, B.S. and Pawar, T.B., 2020. Investigation of Structural and Spectroscopic Parameters of Ethyl 4-(4-isopropylphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate: a DFT Study. Chemistry & Biology Interface, 10(1).
- Ramazani, Ali, Masoome Sheikhi, and Hooriye Yahyaei. “Molecular Structure, NMR, FMO, MEP and NBO Analysis of Ethyl-(Z)-3-phenyl-2-(5-phenyl-2H-1, 2, 3, 4-tetraazol-2-yl)-2-propenoate Based on HF and DFT Calculations.” Chemical Methodologies 1, no. 1 (2017): 28-48.
CrossRef
- Adole, V.A., Jagdale, B.S., Pawar, T.B. and Desale, B.S., 2020. Molecular structure, frontier molecular orbitals, MESP and UV–visible spectroscopy studies of Ethyl 4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate: A theoretical and experimental appraisal. Material Science Research India, 17(specialissue2020), pp.13-36.
CrossRef
- Pathade, S.S. and Jagdale, B.S., 2020. Experimental and Computational Investigations on the Molecular Structure, Vibrational Spectra, Electronic Properties, FMO and MEP Analyses of 4, 6-Bis (4-Fluorophenyl)-5, 6-dihydropyrimidin-2 (1H)-one: A DFT Insight. Physical Chemistry Research, 8(4), pp.671-687.
- Wang, Y., Liu, Q., Qiu, L., Wang, T., Yuan, H., Lin, J. and Luo, S., 2015. Molecular structure, IR spectra, and chemical reactivity of cisplatin and transplatin: DFT studies, basis set effect and solvent effect. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 150, pp.902-908.
CrossRef
- Adole, V.A., Waghchaure, R.H., Jagdale, B.S., Pawar, T.B. and Pathade, S.S., Molecular tructure, frontier molecular orbital and spectroscopic examination on dihydropyrimidinones: a comparative computational approach Journal of Advanced Scientific Research, 2020. 11 (2), pp.64-70.
- Mebi, C.A., 2011. DFT study on structure, electronic properties, and reactivity of cis-isomers of [(NC5H4-S)2Fe(CO)2]. Journal of Chemical Sciences, 123(5), pp.727-731.
CrossRef
- Adole, V.A., Pawar, T.B. and Jagdale, B.S., 2021. DFT computational insights into structural, electronic and spectroscopic parameters of 2-(2-Hydrazineyl) thiazole derivatives: a concise theoretical and experimental approach. Journal of Sulfur Chemistry,42(2) pp. 131-148.
CrossRef
- Adole, V.A., Koli, P.B., Shinde, R.A. and Shinde, R.S., 2020. Computational Insights on Molecular Structure, Electronic Properties, and Chemical Reactivity of (E)-3-(4-Chlorophenyl)-1-(2-Hydroxyphenyl) Prop-2-en-1-one. Material Science Research India, 17(specialissue2020), pp.41-53.
CrossRef
- Pathade, S.S., Adole, V.A., Jagdale, B.S. and Pawar, T.B., 2020. Molecular structure, electronic, chemical and spectroscopic (UV-visible and IR) studies of 5-(4-chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-pyrazole: combined DFT and experimental exploration. Material Science Research India, 17(specialissue2020), pp.27-40.
- Adole, V.A., Jagdale, B.S., Pawar, T.B. and Sawant, A.B., 2020. Experimental and theoretical exploration on single crystal, structural, and quantum chemical parameters of (E)‐7‐(arylidene)‐1,2,6,7‐tetrahydro‐8H‐indeno[5,4‐b] furan‐8‐one derivatives: A comparative study. Journal of the Chinese Chemical Society, 67(10), pp.1763-1777.
CrossRef
- Pawar, T.B., Jagdale, B.S., Sawant, A.B. and Adole, V.A., 2017. DFT Studies of 2-[(2-substitutedphenyl) carbamoyl] benzoic acids. Journal of Chemical, Biological and Physical Sciences, 7, pp.167-175.
- Adole,V.A., Bagul, V.R., Ahire, S.A., Pawar, R.K., Yelmame, G.B.,and Bukane, A.R.,2021. Computational chemistry: molecular structure, spectroscopic (UV-visible and IR), electronic, chemical and thermochemical analysis of 3′-phenyl-1,2- dihydrospiro[indeno[5,4-b], Journal of Advanced Scientific Research, 12(1) Suppl 1, pp. 276-286.
- Frisch, M., Trucks, G., Schlegel, H., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery Jr, J.A., Vreven, T.K.K.N., Kudin, K.N., Burant, J.C. and Millam, J.M., 2004. Gaussian 03, revision C. 02.
- Szafran, M., Katrusiak, A., Koput, J. and Dega-Szafran, Z., 2007. X-ray, MP2 and DFT studies of the structure, vibrational and NMR spectra of homarine. Journal of Molecular Structure, 846(1-3), pp.1-12.
CrossRef
- Dereli, Ö., Sudha, S. and Sundaraganesan, N., 2011. Molecular structure and vibrational spectra of 4-phenylsemicarbazide by density functional method. Journal of Molecular Structure, 994(1-3), pp.379-386.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.