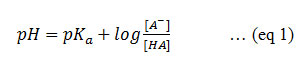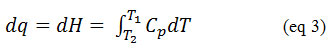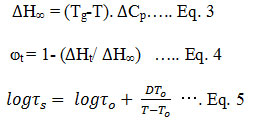Article Publishing History Article Received on : 02 May 2021 Article Accepted on : 09 Jun 2021 Article Published : 15 Jun 2021 Plagiarism Check: Yes Reviewed by: Dr. Paroma Arefin Second Review by: Dr. Sapana Jadoun Final Approval by: Dr. G. Rrajarajan
Article Metrics
ABSTRACT:
In recent years, solid form screening has become an integral and mandatory part of drug development. Solid form screening typically involves producing and characterizingmaximum possible solid forms of a potential drug candidate. Different types of solid forms for future drug product development includes salt screening, co-crystal screening, crystallization process development, polymorph screening as well as amorphous solid dispersion screening.Screening studies of a solid form is a set of carefully designed experiments that requires use of advanced analytical techniques to collect analytical data followed by a thoughtful data analysis.This solid form screening studies guide an important decision-making of lead solid form whichis likely to play a vital role during the pharmaceutical product development lifecycle. The selection criteria include pharmaceutically relevant properties, such as therapeutic efficacy and processing characteristics as well as role of physicochemical properties (i.e. solubility, dissolution rate, hygroscopicity, physical stability and chemical purity) in drug product development. A selected solid form, if thermodynamically unstable, it may undergo solid form changes upon exposure to environmental conditions such as temperature and relative humidity as well as manufacturing stress during the pharmaceutical unit operations. In thepresent work, fundamentals of solid form screening are discussed, including the experimental screening methodologies as well as characterization and analysis of solid forms. The importance of drug product risk assessment pertaining to the desired solid form are also discussed here.
KEYWORDS:
Crystallization; Cocrystal; New Drug Candidate; Polymorph; Screening; Salt; Solid Form
Copy the following to cite this article:
Patel N. A. A Review on Significance of Identifying an Appropriate Solid Form During Drug Discovery and Product Development. Mat. Sci. Res. India;18(2).
Copy the following to cite this URL:
Patel N. A. A Review on Significance of Identifying an Appropriate Solid Form During Drug Discovery and Product Development. Mat. Sci. Res. India;18(2). Available from: https://bit.ly/3gDI0Jj
Introduction
Identifying a thermodynamically stable and acceptable solid form is an essential part that follows drug discovery. In past, overall goal of finding appropriate solid form was achieved by selecting low energy crystalline forms such as salts and polymorphs.1-5 However, the discovery of more poorly soluble drugs has obligated the scientists to explore more solid forms which can be used to improve therapeutic efficacy as well as minimal risk during manufacturing of the drug substance as well as the drug product. Co-crystals and amorphous solid dispersions (ASD) have been emphasized upon in past two decades. Hygroscopicity, physicochemical stability and drugability are other important challenges that needs to be overcome using a solid form with desirable properties.
The dispersal of research activities from early stage to marketing approval depends on several factors.However,cost effectiveness and short timeframe are limiting factors for the researchers in the early stage research and development to identify a relatively stable solid form.6 These challenges often require an effective solid form selection strategy which may explore various aspects of the physicochemical properties of a new chemical moiety.7
Solid form selection requires amulti-testing process.The screening activities in early stagehave become more promisingthrough the availability of automated screening machines, advanced solid-state characterization techniques, and availability of computational approaches.7However the time-sensitivity during early development only allows a limited screening experiments with a complete focus on searching for anappropriate solid form andswift transition to the next stage of pharmacokinetic and pharmacodynamic studies, toxicity studies followed by a preformulation studies.8Once the clinical efficacy is established during the later developmental phase, material availability improves due to scale-up of materialas well as availability of other resources.At this stage, a more comprehensive screens have become more promising through the availability of automated screening machines, advanced solid-state characterization techniques, and availability of computational approaches.9 This exhaustive screening not only allows finding all possiblepolymorphs, but alsoprovide intellectual property protection and demonstrate readiness of optimal solid form for large scale production through a robust crystallization process.10-11
One of the major challenges with newly discovered drugs is their poor solubility. Poorly soluble drug in early drug development poses may lead to incomplete optimization along with increased timelines.12-13 Application of an appropriate solid form screening supported with predicted as well as experimental results expands the possibility of identifying a more soluble form with confidence. Solubility of a drug molecule in aqueous conditions mimicking bodily fluids is an important requirement to determine molecule’s success with increase bioavailability. 1-3,14 If the solubility of a newly discovered solid form is high then it can speed up the preclinical as well as clinical studies. Furthermore, highly soluble drugs may not require time-consuming and complicated advanced enabling technologies to develop a drug product.
Advanced technologies and automation globally have also benefitted solid form screening approaches which not only supports the preparation of samples but also enable in-situ characterization of the sample.15-16Accessibility of these technologies has made evaluation of different solid forms efficient and less time consuming with availability of very limited material in early developmental stages. These activities can be executed in parallel with pharmacokinetics (PK) studies of multiple compounds during lead optimization and enables a rapid decision-making process.2
The overall goal of this review work is to emphasize on a rational that supports a “fit-for-purpose” strategy to employ a successful solid form screening and selection.Furthermore, it is important for a pharmaceutical company to be able to advance a drug candidate to the next stage in cost-effective manner right from the discovery stage à pre-clinical stage à clinical stage à regulatory approval à marketing authorization. In addition to this, the key considerations such as cost, timelines, and product quality and their impact are highlighted during solid form selection.
Physicochemical Properties of a New Drug Candidate
The performance of a drug candidate is largely dependent on its solid form.The physicochemical properties of a solid form such as melting point, solubility, stability, hygroscopicity, and bulk density can have a major influence of its in vivo behavior. Therefore, in the early developmental stage, majority of the efforts are focused on improving the physical properties to enhance the drug processibility, improve absorption, and optimize delivery options.17
Generating pH solubility profile and kinetic as well as equilibrium solubility measurement in different biorelevant media are among the important data that a researcher needs to obtain during the preformulation stage.In-vitro permeability studies using CaCO2 cell membrane is also another important piece of information in early developmental stage. Solubility and permeability data are then used to determine the projected doses for toxicity, pre-clinical and clinical studies.
In addition to the experimental studies, computational results also provide some insights on the physicochemical and invivo behavior of a drug candidate:
Determine
possibility of salt formation by calculating the acid-base dissociation
constant (pKa) value(s)
Based
on solubility and permeability assigning aBiopharmaceutics Classification
System (BCS) to a drug candidate
Calculate
the dose number
Predicting
the possibility of improved physical and chemical stability followed by solid
form selection
Drug product risk assessment strategies during
formulation development
Design solid form screening and formulation strategies18
Biopharmaceutical Considerations
In recent years, 90 % of the newly discovered small molecules suffers from limited solubility. With the detailed understanding of biopharmaceutics, pharmacokinetics and pharmacodynamics of the newly discovered drugs, different classification systems have evolved over the period of time.
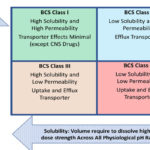
Biopharmaceutics Classification System (BCS)18 (Figure 1),
Developability Classification System (DCS)19(Figure 2) and
Biopharmaceutics Drug Disposition Classification System (BDDCS)20 (Figure 3).
Most of the newly discovered drugs fall into the BCS 2 or 4 category (Figure 1). The poorly soluble compounds have non-linear dose proportionality, limited toxicological coverage, and inter-subject pharmacokinetic variabilities. These challenges may often affect human dosing prediction and future studies.
Therefore, the application of enabling technologies to improve solubility and permeability via extensive solid-form screening and novel drug delivery systems becomes very important to achieve an overall of goalof getting complete toxicological information, followed by aconsistent exposure in different animal species as well as humans as the molecules progresses during the drug life cycle.
Researchers’ main goal is to identify a promising drug candidate for further study in the lab and in animal models, and then in people. Therefore, it is very critical to identify and select a crystalline form that is re-producible, easily processable,as well as chemically pure. These three
characteristics will ensure the availability of drug substance with desired physical properties with a robust process. The availability of drug substance will expedite formulation research to support preclinical and clinical studies.
Salt Screening
As compared to a free form a drug candidate, forming salt using a suitable counterion via salt screening is by far the most effective approach to enhance the solubility of a molecule. Salt formation occurs when a compound is ionized in solution which forms a strong ionic interaction with an oppositely charged counterion.21 From the literature, the pKa difference of more than three between a drug molecule and a counterion may be an underlying mechanism for salt formation.1, 22 The quantum mechanics and interaction between a drug molecule and a counterion are extensively studied even today.9, 23 The salt formation phenomenon improves crystallization of a particular drug candidate. It is well known that salts readily undergo crystallization which further aids in the subsequent processing. 50 % drug products available on market comprises of salt forms. One of the reasons for salt selection includedsolubility improve as well as other physical properties including stability, hygroscopicity and manufacturability.24-26 pKa can be calculated by Henderson Hasselbach equation (eq. 1).
Where, Kais acid dissociation constant, [A–] is concentration of the conjugate base of the acid and [HA] is a concentration of chemical species HA.
For example, levothyroxine sodium, indicated for hypothyroidism, is available for several delivery methods in order to achieve the necessary pharmacokinetic profiles.27 However, levothyroxine sodium is narrow therapeutic index drug which is rate limiting step to achieve desirable therapeutic efficacy. While the sodium salt has a relatively higher solubility than the free acid, this molecule exhibits physicochemical instability. The drug maker developed a tablet and with the efforts from regulatory agency to control the dose variability from dose to dose, the assay specifications have been tightened so as to prevent in vivo variability of levothyroxine sodium.28 While levothyroxine sodium is one example demonstrating the importance of thorough investigation during drug product development. Metformin hydrochloride is another example which has higher solubility as compared to its free form, but it exhibits poor compressibility profiles which available as both immediate release as well as extended-release tablets, which allows the controlled release of a molecule to improve patient compliance.29 Therefore, if a salt exhibits high solubility than the other parameters like physical-chemical instability as well as processability should also be taken into consideration and evaluated accordingly during the developmental stages to prevent any complications during the commercialization phase.
If a poorly soluble compound is ionizable and has potential for salt formation, an extensive salt screening should be employed. A highly soluble and stable salt will aid in minimizing PK variations (inter-subject, inter-species, and dose-to-dose) as it would increase in vivo exposure as well as toxicological coverage. Simple formulations, such as powder in bottle (PiB), powder in capsule (PiC), and suspensions can be easily prepared using a salt form for preclinical and clinical studies. The salt screening of compounds with solubility limitations might be focused on finding a more soluble salt using counter-ions, acetic acid, methanesulfonic acid, citric acid and other such counterions may be preferred ones due to their low molar mass and hydrophilic nature. While improving solubility is a primary goal from salt screening, it also important to recognize more studies may be required as bioavailability also depends on dissolution, precipitation kinetics, and PK studies.30-33It should be recognized that salt formation requires a carefully designed dissolution method to predict the invivo performance of the drug.31-32, 34-35Overall scientific considerations leads to an appropriate decision making in selection of a right salt.
Co-crystal Screening
Co-crystal is a multi-component system that includes a drug and one or more co-formers crystallized into a single crystal lattice.36-37Co-crystal formation can occur between a free form and a co-former, or a salt and a co former. Contrary to the salt formation, co-crystal formation is usually possible between a drug and conformer with a pKa difference less than three. Co- crystallization extends a promising crystal engineering approach to modify a crystal structure and improve physical properties of a given drug.38-39 There are numerous examples drug formulation and manufacturing wherein co-crystallization has offered solutions to the existing problems.38, 40 Due to these advantage, co-crystallization have become an attractive alternative to develop and advance a new chemical entity to the next developmental stage. Novartis’ Entresto®is a co-crystal of two different drugs (i.e. sacubitril and valsartan).This co-crystal not only offers a combination therapy but also exhibits more efficacy than administered individually.40-42 Recent investigations also demonstrate the cocrystals also have improved physical stability as well as mechanical properties, which improves the tabletability of a drug.43-45 Furthermore, size reduction of a tablet is also achievable by use of cocrystal.46-47
Co-crystal screening and salt screening exhibits similarity in multiple ways.48-50 However, at molecular level, co-crystal formation is driven primarily by the H-bonding and other molecular (π – π) interactions between drug and co-former.39, 51-52 Theseinteractions are relatively weak compared to ionic interactions observed in salts. Successful co-crystal screening is dependent on 1) in-depth understanding of structure-property relationship, b) solubility of both API and co-former, and c) specialized screening methods, such as solvent-drop grinding (SDG), homogenous crystallization and thermal methods.37, 53-54At the same time, insufficient knowledge base and limited screening technologies probes challengesfor further use during early developmental stage.42, 55-56
Discovery and identification of few co-crystals,commercial scale production and formulating these cocrystal into drug products may be relatively straightforward as compared to its salt counterpart.57-59 Like any other crystalline material (free form or salt), most co-crystals also require significant efforts in termsco-crystallization process development at large-scale followed stability assessment and biopharmaceutical characteristics assessment in drug products.50, 60-61Approved co-crystal drug products are a clear demonstration of how co-crystal formation can be rewarding, despite theassociated challenges for specific compounds that have limited solubility, poor compressibility and difficult to crystallize out as a free form.62-63With a thorough understanding of co-crystal at molecular level and carefullydesigned efficient screening methodologies will certainlyallow co-crystal to be a more imperative tool during drug development.53, 64-66
Polymorph Screening
Polymorphs are often defined as crystalline forms that have same chemical formula but diverse molecular arrangements and/or conformations within the same crystal lattice.67 Different polymorphs are anticipated to have different physicochemical properties including solubility, stability, micromeritic and mechanical properties.68-69The goal of polymorph screening is to identify different polymorphs, hydrates and solvates.70-71 Importantly, from the polymorph screening studies, it is expected to come up with a form phase map to determine their thermodynamic relationship.72-73 Towards the end of screening studies, the researchers must recommend the most suitable and thermodynamically stable polymorph closer to ambient environmental conditions for further development.6, 11, 74-75Polymorphs screening experiments are generally conducted using small amounts of materials (~30 mg per experiment).An effective screening experimental design should include usingdifferent solvents (polarity, H-bonding donor and acceptor), aqueous solvent mixtures of different water activities, slurry ripening, crystallization conditions, such as temperature and cooling rate, anti-solvent addition, liquid vapor diffusion, solid vapor diffusion, slow evaporation, polymer induced crystallization experimentsas well as computational tools which should be able to investigate various parameters influencing nucleation and growth kinetics of different crystalline forms.7, 76-78. Ideal mole fraction solubilities of a given crystal form can be calculated using the ideal solubility equation (eq 2) using Tm and enthalpy values from DSC measurements.
Where, X is the mole fraction, ΔH is the change in enthalpy, R is the gas constant, Tm is the melting point and T is the temperature at which the ideal solubility needs to be calculated.
While
conducting a polymorph screening, it is very important to include the real-time
conditions during process chemistry, crystallization as well as drug product
process in the screening design so as to identify any risks that may be
encountered drug isolation as well as formulation prior to selection of an
optimal polymorph proposed for preclinical and clinical studies. Process-induced
solid form transformation is equally important during drug product unit
operations including API micronization, wet granulation, tableting, and solid-state
interaction with excipients.79-81
Polymorphs can be divided into two categories, enantiotropic polymorphs and monotropic polymorphs. The relatively stable form can be determined by Berger-Ram Berger Rule using enthalpy of fusion and melting temperature results from differential scanning calorimetry (DSC), competitive slurry experiments and Van’t Hoff plot by obtaining solubility at different temperatures.
The heat flow (dq) in the DSC heating a sample is equal a change in enthalpy. A change in enthalpy is further related to the heat capacity Cp, as described in eq. 3, 82
Where, dqis the heat transferred, Cpis the heat capacity, T is the temperature.
Researchers can imagine how difficult it can be to switch to a different polymorphic form at a later stage.83-84 These changes at a later stage will likely invite additional work. These activities may include crystallization process development, reformulation, as well as possibly bridging toxicology and PK studies. This could lead to significant delays in drug product life cycle as well as high costs.
The
International Conference on Harmonization (ICH) guidelines mandates polymorph
screening for a new drug moleculeto be included in the chemistry,
manufacturing, and controls (CMC) section of a regulatory submission. Polymorph
screening gives confidencein the robustness of drug substance as well as drug
product manufacturing processes. Selecting a stable polymorph at ambient
conditions can avoid associated risks with drug product and guarantee that the
drug product is stable, efficacious, and safe for patients.
As the drug candidate advances to a later stage in drug development, a more comprehensive screening may be required depending on the route of administration and drug delivery system which may including amorphous material.
Crystallization Process Development
One a desired solid form is selected a robust crystallization process is required to isolate the solid form at a commercial scale with a potential for impurity rejection.84 This step becomes even more critical especially for high-volume drug products, wherein,cost of a drug substance contributes to the overall cost of drug product.85-87A successful final drug crystallization process requires finding solvents system through series of metastable zone width solubility experiment to determine a phase boundary within practical ranges of critical processing parameters.88-91 Finally, a developed and optimized process should meet requirements and specifications of particle attributes with controlled particle size distribution (PSD), crystal morphology or shape, bulk density and offers yield maximization with thorough understanding of the thermodynamics and kinetics of a specific (polymorphs/solvents) system.10 With recent advances in crystallization process development, a real-time in-process information can be continuously monitored and analyzed using in situ process analytical technologies (PATs).10, 86, 92 This will provide a better understanding and control of crystallization processes. Some of PATs used nowadays include Focused beam reflectance measurement, BlazeMetrics®, process Raman, and ReactI Rapplied by scientist during process development.10, 93-94 A suitable crystalline form of a drug candidate is important in determining the success of manufacturing on a large commercial scale due to the aforementioned benefits a crystalline form can offer by ensuring product quality and safety.10
Amorphous Solid Dispersion Screening
The drug candidates for which it is challenging to obtain a stable crystalline form with acceptable solubilities will require enabling technologies to improve solubility studies. Amorphous solid dispersions (ASD) are often explored and used to improve solubility for compounds with limited solubility. Like salts, co-crystal and polymorph selection, an amorphous dispersion also required a detailed screening protocol to search a suitable amorphous dispersion with desirable properties. The main goal of amorphous solid dispersions is to improvea higher kinetic solubility for better drug absorption compared to its crystalline counterpart. The expected solubility improvement of an amorphous solid typically ranges from approximately 2 to 1,000 folds.25 Although, the presence of disorder in the amorphous solids complicates the system, the crystalline lattice energy and H-bonding donors and acceptors ina molecule do offer insights on the predicted physicochemical properties improvement.95-97
The ASD requires optimization of polymers and surfactants which can inhibit precipitation and re-crystallization of the drug in supersaturated solutions.98-100Different methodologies are employed to prepare ASD, such as rapid solvent evaporation, spray drying (SD) of solutions, freeze-drying (FD), or hot melt extrusion (HME).25, 101-103With the increase in demand of ASDs due to discovery of poorly soluble compounds have forced the equipment manufacturers to provide machineries which can be used to test these technologies at very small scale (as low as ~10 g material). These technologies can be easily scaled up to suffice large scale production requirements during commercialization state.104 One of the most challenging problem associated with ASD is its physical stability and risk of recrystallization to the most stable form. Efforts have been made to study and predict the stability of ASD. Zografi et al has reported that if the ASD is stored at a temperature 50 °C below glass transition temperature then the recrystallization kinetics can be slowed down significantly. The physical stability of ASD is dependent on maximum enthalpic recovery (eq. 3), extent of relaxation (eq. 4) and relaxation time (Eq. 5). 105
Where, ∆H∞is the maximum enthalpic recovery, Tg is a glass transition temperature at a given temperature T, Cpis the heat capacity, ⱷt is the extent of relaxation, τs is structural relaxation time, D is the fragility parameter and Toi s the initial temperature. The ASD formulation requires screening in early development that includes testing solubility of drug in organic solvents suitable for solid dispersion, drug-polymermiscibility, and drug interaction with surfactants. Drug loading plays a major role in deciding the formulation for ASD preparation. Advanced solid-state characterization techniques including polarized light microscopy, Xray powder diffraction, differential scanning calorimetry, thermogravimetric analysis, dissolution profiles, and accelerated stability assessment are required to characterize ASD formulations. The promising leadsamong ASD formulations arefurther tested for PK studies in animal to gain confidence on the selected ASD for further development.
Formulation Strategy and Drug Delivery τs
Solid form selection is directly dependent on various drug product development aspects including route of administration, dose, dosage form, and release profile of a drug. Early product development support highlights the role of solid form selection for pre clinical and clinical studies.A relatively stable solid form is typically moved on to the final stageto achievero bust manufacturing of commercial formulation. Ahighly soluble salt is a preferred choice for a formulation meant for oral delivery. Contradictorily, a free form which isunionized may be chosen for topical applications wherein permeability of a drug is more important parameter as compared to solubility. Each drug delivery system may have a specific preference in terms of physicochemical properties as well as desired solid form. Solid-state stability, adequate solubility and dissolution rate, drug-excipient compatibility, bulk density, micromeritics, and compressibility are few among the important consideration for an oral drug delivery system. It should be noted that highly soluble salt in aqueous systemsisnot direct outcome for bioavailability.A relatively stable salt form in solid-state may undergo precipitation as well as disproportionationin vivo, which might hamper the bioavailability for that particular drug.35, 106 In such cases, polymeric excipients as well as surfactants must be included in the formulation to inhibit the nucleation and crystallization of the free form. This should be thoroughly investigated during the early developmental stages to avoid patient-based failure modes in future.33 As in the inhalation formulations, physicochemical compatibility with excipients as well as device components, hygroscopicity, and milling are specifically important. Liquid formulations such as solutions or suspensions areconventionally used to carry out toxicological studies. The similar approach is used for parental formulations, intranasal, and pulmonary delivery systems. In these drug delivery systems, the drug is in direct contact with formulation vehicle either solubilized or suspended which is also crucial while selecting the solid form.While considering salt as a solid form for parenteral dosage, the pH of the salt solution at the desired concentration in the biorelevant medium should be investigated to assess the acceptability of pH compatibility with physiological fluids. In addition to this, solubilized formulations needs apH solubility profile knowledge databaseso to ensure the drug concentration is well below the equilibrium solubility of the most stable form in the formulation vehicle to maintain the under-saturated without triggering self-nucleation or crystallization.8In addition to the important aspects discussed above, the solid form selection becomes even more challenging for pediatric formulations.107-109
Solid form Screening and Selection Strategies
Each newly discovered drug candidate has a specific physicochemical and quantomechanical properties which requires careful application of solid-form strategy, which typically varies from molecule to molecule during the drug development life cycle.Table 1 delineates the application of solid form investigations aligned with developmental stages to achieve phase-specific developmental goals. Early developmental stages focus on searching a suitable crystalline form.Crystalline solid formenables availability of improved isolation process as well as chemically pure drug substance.Highly soluble and physic-chemically stable drug substance can be further used in preclinical, toxicity and clinical studies. As the drug substance progresses further with supportive data from toxicity studies, further studies are carried out during the later stages to evaluate the solid form risks associated with drug substance and drug product manufacturing. These studies may include an extensive polymorph screening to establish a form phase relationship map.This would maximize opportunities to advance the molecule with new indications and novel formulations to the final and commercial stage of drug product life cycle.
Table 1: Solid form investigations at each developmental stage during drug product life cycle.
Drug Product Lifecycle Stage
Key Development Objective
Solid Form Investigations
Discovery
(lead to candidate selection)
Selection of a crystalline form for isolation and purification purposes (anhydrates, hydrate and solvates)110
Crystallization, salt / co-crystal screening
Early Development
(PK, Tox, Phase 1 and 2a)
Assess polymorphism and chose the most stable solid form at ambient conditions
ASD to the exposure of poorly soluble drugs
Polymorph screening and selection
Revisit salt/ co-crystal screening
ASD screening
Late Development
(Phase 2b and 3, Launch)
The optimal solid form to support pivotal study till commercial launch
Finalize drug substance as well as drug product manufacturing process
Comprehensive polymorph screening
Process risk assessment and mitigation
Drug product risk assessment
Life Cycle Management
(New Indications & formulations)
Comprehensive solid form knowledge
Optimal form of new drug manufacturing, new indications, and novel formulations
Comprehensive salt / co-crystal screening
Bridging Drug Discovery and Formulation Development
Drug product life cycle is generally divided into two stages. First stage involves discovery of drug with the help from medicinal chemists whose focus is to develop a chemical moiety with desired therapeutic effect through multiple organic reactions as well as structure activity relationships knowledge. Once the molecule is deemed effective against a specific biological target that is important in a disease, it progresses to the next stage. Stage two involves the placement of a drug molecule into a drug delivery system by formulation scientists to investigate the safety and efficacy of the newly discovered molecule. The transition from stage one to stage two can sometimes be very challenging.
Medicinal chemists invest their knowledge and time towards optimizing a lead molecule with best selectivity and in vitro potency. At the discovery stage, the focus of the studies lies on in vitro potency of candidate molecules, which typically has poor physicochemical and biopharmaceutical properties. There is neither any understanding on non-clinical as well as clinical dosing nor the formulation and dosage form technologies that may be needed in future.
Therefore, it is vital to transition the lead molecule from the hands of medicinal chemists to the preformulation and formulation scientists who understands the physicochemical as well as biopharmaceutical importance of a potential drug candidate. The role and involvement of introducing formulation scientists during discovery stage to next stage is very diverse among different companies. In general, it is towards the late discovery stage, wherein, a particular salt and desired polymorph has been decided.
A disconnect in the transition of lead molecule is clearly observed in this approach conventionally that most biotech companies follow. The primary focusemphasizes on resolving the solubility issues, wherein other important parameters could be ignored, instead of putting all resources to get a 360° view on the overall characteristics of a potential drug molecule.
A recent review work demonstrated that salts were selected in the early developmental stages due to ease of synthesis and crystallization and economic viability. The same salt forms had no or minimal understanding on the downstream processes (physical and chemical stability, process ability into dosage forms, solubility, and dissolution rate at different pH conditions). Hypothetically, if the lead salt form does not have desired solid-form properties, it may become very difficult to alter the salt form during the later stages. If at all, a different salt is selected for further development then it would significantly increase the timelines as well as costs. All the important studies such as biological, toxicological, formulation, and stability tests may need to be revisited using a new salt form. This ultimately led to longer timelines and increased costs. 24
Conclusion
The solid form of a potential drug candidatemight have a deep impact on the physicochemical properties and drug developmental activities. The solid form selection strategy is outlined in this review work. The current work highlights the fundamental consideration of molecule’s physicochemical properties, the role of different solid forms in rejecting undesired impurities physical properties, as well formulation approaches. This can provide a framework to develop an appropriate solid form selection strategy of small molecule drug candidates wherein the drug candidate advances to the next developmental stage rapidly. Importantly, it should be noted that the lack of detailed understanding of a solid form at molecular level has resulted in a drug product recall due to pharmaceutical quality concerns. This can be very well exemplified by the most recent drug product recall of levothyroxine sodium tablets manufactured by Acella Pharmaceuticals from the US market on Apr 29th, 2021.
Lastly, the researchers should acknowledge that discovering a soluble solid form using an enabling technology to improve the solubility and dissolution of a candidate is just the first step in drug development. The later steps include determination of doses, dosing strategies, devising a drug delivery system and drug product risk assessment which will ensure the complete drug absorption occurs without chemical degradation, physicochemical instability or disproportion ation of the drug in physiological fluids. The safety and efficacy of a drug product can be ensured by availability of new prior knowledge.111
Expert Opinion
Solid form screening has been explored extensively by the researchers in drug product development and it is very well established. The drug properties of a potential new chemical entity can only be understood if the solid-state structure of drugs molecules have been thoroughly studied by itself as well as its impact on the future drug formulation. Each researcher has their own screening workflows, thoughtful strategies, and availability to advanced analytical tools to help choose a stable solid form at ambient conditions. However, the pharmaceutical industry and the regulatory agencies are familiar with the fact that the drug product recalls have happened in past and it has led to evolution of regulatory guidance to streamline the research activities pertaining to solid form selection and enable a possibility of robust drug product on market.This manuscript has summarized different examples that aidthe development programs at pharmaceutical companies for a newly discovered molecule.
Acknowledgment
The author would like to acknowledge Institute of Pharmacy, Nirma University as well as its faculty for providing and extending educational and technical support.
Conflict
Of Interests
Theauthor(s) declare(s) that there is no conflict of interests regarding the publication of thisarticle
Funding
Source
The
author(s) declare(s) that the funding is done by author only.
References
Stahl, P. H.; Wermuth, C. G., Pharmaceutical salts: Properties, selection and use. John wiley & sons: 2002.
Huang, L.-F.; Tong, W.-Q. T., Impact of solid state properties on developability assessment of drug candidates. Advanced Drug Delivery Reviews 2004,56 (3), 321-334.
Ku, M., Salt and polymorph selection strategy based on the biopharmaceutical classification system for early pharmaceutical development. American Pharmaceutical Review 2010,13 (1), 22-30.
Davies, G., Changing the salt, changing the drug. Pharmaceutical Journal 2001,266 (7138), 322-323.
Byrn, S.; Pfeiffer, R.; Ganey, M.; Hoiberg, C.; Poochikian, G., Pharmaceutical solids: a strategic approach to regulatory considerations. Pharmaceutical Research 1995,12 (7), 945-954.
Miller, J. M.; Collman, B. M.; Greene, L. R.; Grant, D. J.; Blackburn, A. C., Identifying the stable polymorph early in the drug discovery–development process. Pharmaceutical Development and Technology 2005,10 (2), 291-297.
Lee, E. H., A practical guide to pharmaceutical polymorph screening & selection. Asian Journal of Pharmaceutical Sciences 2014,9 (4), 163-175.
Al-Achi, A.; Gupta, M. R.; Stagner, W. C., Integrated pharmaceutics: applied preformulation, product design, and regulatory science. John Wiley & Sons: 2013.
Abramov, Y. A., Computational Pharmaceutical Solid‐State Chemistry: An Introduction. Wiley Online Library: 2016.
Simon, L. L.; Simone, E.; Oucherif, K. A., Crystallization process monitoring and control using process analytical technology. In Computer Aided Chemical Engineering, Elsevier: 2018; Vol. 41, pp 215-242.
Beckmann, W.; Otto, W.; Budde, U., Crystallisation of the stable polymorph of hydroxytriendione: seeding process and effects of purity. Organic Process Research & Development 2001,5 (4), 387-392.
Leeson, P. D., Molecular inflation, attrition and the rule of five. Advanced Drug Delivery Reviews 2016,101, 22-33.
Hann, M. M.; Keserü, G. M., Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nature reviews Drug discovery 2012,11 (5), 355-365.
Chemburkar, S. R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W., Dealing with the impact of ritonavir polymorphs on the late stages of bulk drug process development. Organic Process Research & Development 2000,4 (5), 413-417.
Morissette, S. L.; Almarsson, Ö.; Peterson, M. L.; Remenar, J. F.; Read, M. J.; Lemmo, A. V.; Ellis, S.; Cima, M. J.; Gardner, C. R., High-throughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids. Advanced Drug Delivery Reviews 2004,56 (3), 275-300.
Igo, D. H.; Chen, P., Vibrational spectroscopy of solid‐state forms–applications and examples. Handbook of Vibrational Spectroscopy 2006.
Lindenmayer, J., Long-acting injectable antipsychotics: focus on olanzapine pamoate. Neuropsychiatric Disease and Treatment 2010,6, 261.
Amidon, G. L.; Lennernäs, H.; Shah, V. P.; Crison, J. R., A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharmaceutical Research 1995,12 (3), 413-420.
Butler, J. M.; Dressman, J. B., The developability classification system: application of biopharmaceutics concepts to formulation development. Journal of Pharmaceutical Sciences 2010,99 (12), 4940-4954.
Benet, L. Z.; Amidon, G. L.; Barends, D. M.; Lennernäs, H.; Polli, J. E.; Shah, V. P.; Stavchansky, S. A.; Lawrence, X. Y., The use of BDDCS in classifying the permeability of marketed drugs. Pharmaceutical Research 2008,25 (3), 483-488.
Saal, C.; Becker, A., Pharmaceutical salts: A summary on doses of salt formers from the Orange Book. European Journal of Pharmaceutical Sciences 2013,49 (4), 614-623.
Hilal, S.; El-Shabrawy, Y.; Carreira, L.; Karickhoff, S.; Toubar, S.; Rizk, M., Estimation of the ionization pKa of pharmaceutical substances using the computer program Sparc. Talanta 1996,43 (4), 607-619.
Vepuri, S. B.; Devarajegowda, H.; Soliman, M. E., Synthesis, characterization and molecular modelling of a novel dipyridamole supramolecule–X-ray structure, quantum mechanics and molecular dynamics study to comprehend the hydrogen bond structure–activity relationship. Journal of Molecular Structure 2016,1105, 194-204.
Serajuddin, A. T., Salt formation to improve drug solubility. Advanced Drug Delivery Reviews 2007,59 (7), 603-616.
Shah, H.; Shah, V.; Parikh, D.; Butani, S.; Mehta, T., Dissolution improvement of nebivolol hydrochloride using solid dispersion adsorbate technique. Asian Journal of Pharmaceutics 2015,9 (1), 49-55.
Surov, A. O.; Vasilev, N. A.; Voronin, A. P.; Churakov, A. V.; Emmerling, F.; Perlovich, G. L., Ciprofloxacin salts with benzoic acid derivatives: structural aspects, solid-state properties and solubility performance. CrystEngComm 2020,22 (25), 4238-4249.
Shah, H. S.; Chaturvedi, K.; Hamad, M.; Bates, S.; Hussain, A.; Morris, K., New Insights on Solid-State Changes in the Levothyroxine Sodium Pentahydrate during Dehydration and its Relationship to Chemical Instability. AAPS PharmSciTech 2019,20 (1), 39.
USFDA Drug Enforcement Report. https://www.accessdata.fda.gov/scripts/ires/index.cfm#tabNav_advancedSearch.
Erdemir, D.; Rosenbaum, T.; Chang, S.-Y.; Wong, B.; Kientzler, D.; Wang, S.; Desai, D.; Kiang, S., Novel co-processing methodology to enable direct compression of a poorly compressible, highly water-soluble active pharmaceutical ingredient for controlled release. Organic Process Research & Development 2018,22 (10), 1383-1392.
Jamil, R.; Xu, T.; Shah, H. S.; Adhikari, A.; Sardhara, R.; Nahar, K.; Morris, K. R.; Polli, J. E., Similarity of dissolution profiles from biorelevant media: Assessment of interday repeatability, interanalyst repeatability, and interlaboratory reproducibility using ibuprofen and ketoconazole tablets. European Journal of Pharmaceutical Sciences 2021,156, 105573.
Chaturvedi, K.; Shah, H. S.; Sardhara, R.; Nahar, K.; Dave, R. H.; Morris, K. R., Protocol development, validation, and troubleshooting of in-situ fiber optic bathless dissolution system (FODS) for a pharmaceutical drug testing. Journal of Pharmaceutical and Biomedical Analysis 2021,195, 113833.
Shah, H. S.; Sardhara, R.; Nahar, K.; Xu, T.; Delvadia, P.; Siddiqui, A.; Gao, Z.; Selen, A.; Morris, K., Development and Validation of Sample Preparation and an HPLC Analytical Method for Dissolution Testing in Fed-State Simulated Gastric Fluid—Illustrating Its Application for Ibuprofen and Ketoconazole Immediate Release Tablets. AAPS PharmSciTech 2020,21 (5), 1-13.
Shah, H. S. Understanding and Classifying the Solid-State Properties of Selected Narrow Therapeutic Index Drug Substances and Modeling the Contribution of Stress Induced Changes on Drug Product Failure Modes. Long Island University, The Brooklyn Center, 2019.
Sardhara, R.; Chaturvedi, K.; Shah, H. S.; Vinjamuri, B. P.; Al-Achi, A.; Morris, K. R.; Haware, R. V., Predictive Performance Comparison of Computed Linear and Quadratic Multivariate Models for In-Situ UV Fiber Optics Tablet Dissolution Testing. European Journal of Pharmaceutical Sciences 2021,161, 105806.
Shah, H. S.; Chaturvedi, K.; Dave, R. H.; Morris, K. R., Molecular Insights into Warfarin Sodium 2-Propanol Solvate Solid Form Changes and Disproportionation Using a Low Volume Two-Stage Dissolution Approach. Molecular Pharmaceutics 2021,18 (4), 1779-1791.
Sun, C. C., Cocrystallization for successful drug delivery. Expert Opinion on Drug Delivery 2013,10 (2), 201-213.
Wang, C.; Sun, C. C., The landscape of mechanical properties of molecular crystals. CrystEngComm 2020,22 (7), 1149-1153.
Perumalla, S. R.; Sun, C. C., Improved solid-state stability of salts by cocrystallization between conjugate acid–base pairs. CrystEngComm 2013,15 (29), 5756-5759.
Vasilev, N. A.; Surov, A. O.; Voronin, A. P.; Drozd, K. V.; Perlovich, G. L., Novel cocrystals of itraconazole: Insights from phase diagrams, formation thermodynamics and solubility. International Journal of Pharmaceutics 2021,599, 120441.
Babu, N. J.; Nangia, A., Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Crystal Growth & Design 2011,11 (7), 2662-2679.
Bysouth, S. R.; Bis, J. A.; Igo, D., Cocrystallization via planetary milling: Enhancing throughput of solid-state screening methods. International Journal of Pharmaceutics 2011,411 (1-2), 169-171.
Kumar, S., Pharmaceutical cocrystals: an overview. Indian Journal of Pharmaceutical Sciences 2018,79 (6), 858-871.
Joshi, T. V.; Singaraju, A. B.; Shah, H. S.; Morris, K. R.; Stevens, L. L.; Haware, R. V., Structure–mechanics and compressibility profile study of flufenamic acid: nicotinamide cocrystal. Crystal Growth & Design 2018,18 (10), 5853-5865.
Sun, C. C.; Hou, H., Improving mechanical properties of caffeine and methyl gallate crystals by cocrystallization. Crystal Growth and Design 2008,8 (5), 1575-1579.
Sun, C. C.; Grant, D. J., Improved tableting properties of p-hydroxybenzoic acid by water of crystallization: a molecular insight. Pharmaceutical Research 2004,21 (2), 382-386.
Bhatt, J.; Shah, H.; Basim, P.; Morris, K.; Haware, R. In Structure–Mechanics Study of Cocrystals to Optimize Tablet Size, NIPTE Conference, 2018.
Bhatt, J. A.; Bahl, D.; Morris, K.; Stevens, L. L.; Haware, R. V., Structure-mechanics and improved tableting performance of the drug-drug cocrystal metformin: salicylic acid. European Journal of Pharmaceutics and Biopharmaceutics 2020,153, 23-35.
Aakeröy, C. B.; Fasulo, M. E.; Desper, J., Cocrystal or salt: does it really matter? Molecular Pharmaceutics 2007,4 (3), 317-322.
Childs, S. L.; Stahly, G. P.; Park, A., The salt− cocrystal continuum: the influence of crystal structure on ionization state. Molecular Pharmaceutics 2007,4 (3), 323-338.
Nagapudi, K.; Umanzor, E. Y.; Masui, C., High-throughput screening and scale-up of cocrystals using resonant acoustic mixing. International Journal of Pharmaceutics 2017,521 (1-2), 337-345.
Surov, A. O.; Voronin, A. P.; Vasilev, N. A.; Ilyukhin, A. B.; Perlovich, G. L., Novel cocrystals of the potent 1, 2, 4-thiadiazole-based neuroprotector with carboxylic acids: virtual screening, crystal structures and solubility performance. New Journal of Chemistry 2021,45 (6), 3034-3047.
Jagia, M. Screening, preparation and characterization of novel co-crystals and eutectics of the drug Febuxostat. Long Island University, The Brooklyn Center, 2017.
Musumeci, D.; Hunter, C. A.; Prohens, R.; Scuderi, S.; McCabe, J. F., Virtual cocrystal screening. Chemical Science 2011,2 (5), 883-890.
Lu, E.; Rodríguez-Hornedo, N.; Suryanarayanan, R., A rapid thermal method for cocrystal screening. CrystEngComm 2008,10 (6), 665-668.
Kale, D. P.; Zode, S. S.; Bansal, A. K., Challenges in translational development of pharmaceutical cocrystals. Journal of Pharmaceutical Sciences 2017,106 (2), 457-470.
Vioglio, P. C.; Chierotti, M. R.; Gobetto, R., Pharmaceutical aspects of salt and cocrystal forms of APIs and characterization challenges. Advanced Drug Delivery Reviews 2017,117, 86-110.
Cavanagh, K. L.; Maheshwari, C.; Rodríguez-Hornedo, N., Understanding the differences between cocrystal and salt aqueous solubilities. Journal of Pharmaceutical Sciences 2018,107 (1), 113-120.
Mohamed, S.; Tocher, D. A.; Price, S. L., Computational prediction of salt and cocrystal structures—Does a proton position matter? International Journal of Pharmaceutics 2011,418 (2), 187-198.
Aitipamula, S.; Banerjee, R.; Bansal, A. K.; Biradha, K.; Cheney, M. L.; Choudhury, A. R.; Desiraju, G. R.; Dikundwar, A. G.; Dubey, R.; Duggirala, N., Polymorphs, salts, and cocrystals: what’s in a name? Crystal Growth & Design 2012,12 (5), 2147-2152.
Douroumis, D.; Ross, S. A.; Nokhodchi, A., Advanced methodologies for cocrystal synthesis. Advanced Drug Delivery Reviews 2017,117, 178-195.
Stolar, T.; Lukin, S.; Tireli, M.; Sović, I.; Karadeniz, B.; Kereković, I.; Matijašić, G.; Gretić, M.; Katančić, Z.; Dejanović, I., Control of pharmaceutical cocrystal polymorphism on various scales by mechanochemistry: transfer from the laboratory batch to the large-scale extrusion processing. ACS Sustainable Chemistry & Engineering 2019,7 (7), 7102-7110.
Good, D. J.; Rodriguez-Hornedo, N., Solubility advantage of pharmaceutical cocrystals. Crystal Growth and Design 2009,9 (5), 2252-2264.
Yang, D.; Cao, J.; Jiao, L.; Yang, S.; Zhang, L.; Lu, Y.; Du, G., Solubility and Stability Advantages of a New Cocrystal of Berberine Chloride with Fumaric Acid. ACS Omega 2020,5 (14), 8283-8292.
Khalaji, M.; Potrzebowski, M. J.; Dudek, M. K., Virtual Cocrystal Screening Methods as Tools to Understand the Formation of Pharmaceutical Cocrystals—A Case Study of Linezolid, a Wide-Range Antibacterial Drug. Crystal Growth & Design 2021,21 (4), 2301-2314.
Grecu, T.; Hunter, C. A.; Gardiner, E. J.; McCabe, J. F., Validation of a computational cocrystal prediction tool: comparison of virtual and experimental cocrystal screening results. Crystal Growth & Design 2014,14 (1), 165-171.
Barbas, R.; Font-Bardia, M.; Paradkar, A.; Hunter, C. A.; Prohens, R., Combined Virtual/Experimental Multicomponent Solid Forms Screening of Sildenafil: New Salts, Cocrystals, and Hybrid Salt–Cocrystals. Crystal Growth & Design 2018,18 (12), 7618-7627.
Ford, E. B., Polymorphism. Biological Reviews 1945,20 (2), 73-88.
Jain, A.; Shah, H. S.; Johnson, P. R.; Narang, A. S.; Morris, K. R.; Haware, R. V., Crystal anisotropy explains structure-mechanics impact on tableting performance of flufenamic acid polymorphs. European Journal of Pharmaceutics and Biopharmaceutics 2018,132, 83-92.
Vasilev, N.; Voronin, A.; Surov, A.; Perlovich, G. In Solid Forms of Ciprofloxacin Salicylate: Ppolymorphism, Formation Pathways, and Thermodynamic Stability, Mendeleev, 2019; pp 364-364.
Shah, H. S.; Chaturvedi, K.; Zeller, M.; Bates, S.; Morris, K., A threefold superstructure of the anti-epileptic drug phenytoin sodium as a mixed methanol solvate hydrate. Acta Crystallographica Section C: Structural Chemistry 2019,75 (9), 1213-1219.
Surov, A. O.; Voronin, A. P.; Vasilev, N. A.; Churakov, A. V.; Perlovich, G. L., Cocrystals of Fluconazole with Aromatic Carboxylic Acids: Competition between Anhydrous and Hydrated Solid Forms. Crystal Growth & Design 2019,20 (2), 1218-1228.
Pulido, A.; Chen, L.; Kaczorowski, T.; Holden, D.; Little, M. A.; Chong, S. Y.; Slater, B. J.; McMahon, D. P.; Bonillo, B.; Stackhouse, C. J., Functional materials discovery using energy–structure–function maps. Nature 2017,543 (7647), 657-664.
Aaltonen, J.; Allesø, M.; Mirza, S.; Koradia, V.; Gordon, K. C.; Rantanen, J., Solid form screening–a review. European Journal of Pharmaceutics and Biopharmaceutics 2009,71 (1), 23-37.
Seton, L.; Khamar, D.; Bradshaw, I. J.; Hutcheon, G. A., Solid state forms of theophylline: presenting a new anhydrous polymorph. Crystal Growth & Design 2010,10 (9), 3879-3886.
Jarring, K.; Larsson, T.; Stensland, B.; Ymen, I., Thermodynamic stability and crystal structures for polymorphs and solvates of formoterol fumarate. Journal of Pharmaceutical Sciences 2006,95 (5), 1144-1161.
Alvarez, A. J.; Singh, A.; Myerson, A. S., Polymorph screening: comparing a semi-automated approach with a high throughput method. Crystal Growth and Design 2009,9 (9), 4181-4188.
Gu, C.-H.; Young Jr, V.; Grant, D. J., Polymorph screening: influence of solvents on the rate of solvent-mediated polymorphic transformation. Journal of Pharmaceutical Sciences 2001,90 (11), 1878-1890.
Galek, P. T.; Allen, F. H.; Fábián, L.; Feeder, N., Knowledge-based H-bond prediction to aid experimental polymorph screening. Crystal Engineering and Communications 2009,11 (12), 2634-2639.
Chaturvedi, K.; Gajera, B. Y.; Xu, T.; Shah, H.; Dave, R. H., Influence of processing methods on physico-mechanical properties of Ibuprofen/HPC-SSL formulation. Pharmaceutical Development and Technology 2018, 1-9.
Newman, A.; Wenslow, R., Solid form changes during drug development: good, bad, and ugly case studies. AAPS Open 2016,2 (1), 1-11.
Morris, K. R.; Griesser, U. J.; Eckhardt, C. J.; Stowell, J. G., Theoretical approaches to physical transformations of active pharmaceutical ingredients during manufacturing processes. Advanced Drug Delivery Reviews 2001,48 (1), 91-114.
Dafermos, C. M. The second law of thermodynamics and stability; BROWN UNIV PROVIDENCE RI LEFSCHETZ CENTER FOR DYNAMICAL SYSTEMS: 1978.
Kwokal, A., Preparation, Stabilisation and Advantages of Metastable Polymorphs. In Engineering Crystallography: From Molecule to Crystal to Functional Form, Springer: 2017; pp 247-260.
Mortko, C. J.; Sheth, A. R.; Variankaval, N.; Li, L.; Farrer, B. T., Risk assessment and physicochemical characterization of a metastable dihydrate API phase for intravenous formulation development. Journal of Pharmaceutical Sciences 2010,99 (12), 4973-4981.
Fujiwara, M.; Nagy, Z. K.; Chew, J. W.; Braatz, R. D., First-principles and direct design approaches for the control of pharmaceutical crystallization. Journal of Process Control 2005,15 (5), 493-504.
Togkalidou, T.; Braatz, R. D.; Johnson, B. K.; Davidson, O.; Andrews, A., Experimental design and inferential modeling in pharmaceutical crystallization. AIChE Journal 2001,47 (1), 160-168.
Cote, A.; Erdemir, D.; Girard, K. P.; Green, D. A.; Lovette, M. A.; Sirota, E.; Nere, N. K., Perspectives on the Current State, Challenges, and Opportunities in Pharmaceutical Crystallization Process Development. Crystal Growth & Design 2020,20 (12), 7568-7581.
Kadam, S. S.; Kulkarni, S. A.; Ribera, R. C.; Stankiewicz, A. I.; ter Horst, J. H.; Kramer, H. J., A new view on the metastable zone width during cooling crystallization. Chemical Engineering Science 2012,72, 10-19.
Nagy, Z. K.; Fujiwara, M.; Woo, X. Y.; Braatz, R. D., Determination of the kinetic parameters for the crystallization of paracetamol from water using metastable zone width experiments. Industrial & Engineering Chemistry Research 2008,47 (4), 1245-1252.
Sangwal, K., On the estimation of surface entropy factor, interfacial tension, dissolution enthalpy and metastable zone-width for substances crystallizing from solution. Journal of Crystal Growth 1989,97 (2), 393-405.
O’Grady, D.; Barrett, M.; Casey, E.; Glennon, B., The effect of mixing on the metastable zone width and nucleation kinetics in the anti-solvent crystallization of benzoic acid. Chemical Engineering Research and Design 2007,85 (7), 945-952.
Lawrence, X. Y.; Lionberger, R. A.; Raw, A. S.; D’Costa, R.; Wu, H.; Hussain, A. S., Applications of process analytical technology to crystallization processes. Advanced Drug Delivery Reviews 2004,56 (3), 349-369.
De Beer, T.; Allesø, M.; Goethals, F.; Coppens, A.; Vander Heyden, Y.; Lopez De Diego, H.; Rantanen, J.; Verpoort, F.; Vervaet, C.; Remon, J. P., Implementation of a process analytical technology system in a freeze-drying process using Raman spectroscopy for in-line process monitoring. Analytical Chemistry 2007,79 (21), 7992-8003.
Trampuž, M.; Teslić, D.; Likozar, B., Process analytical technology-based (PAT) model simulations of a combined cooling, seeded and antisolvent crystallization of an active pharmaceutical ingredient (API). Powder Technology 2020,366, 873-890.
Yu, L., Polymorphism in molecular solids: an extraordinary system of red, orange, and yellow crystals. Accounts of Chemical Research 2010,43 (9), 1257-1266.
Chaturvedi, K.; Shah, H. S.; Nahar, K.; Dave, R.; Morris, K. R., Contribution of Crystal Lattice Energy on the Dissolution Behavior of Eutectic Solid Dispersions. ACS Omega 2020,5 (17), 9690-9701.
Kaffash, E.; Badiee, A.; Akhgari, A.; Rezayat, N. A.; Abbaspour, M.; Saremnejad, F., Development and characterization of a multiparticulate drug delivery system containing indomethacin-phospholipid complex to improve dissolution rate. Journal of Drug Delivery Science and Technology 2019,53, 101177.
Gumaste, S. G.; Gupta, S. S.; Serajuddin, A. T., Investigation of polymer-surfactant and polymer-drug-surfactant miscibility for solid dispersion. The AAPS Journal 2016,18 (5), 1131-1143.
Chen, J.; Ormes, J. D.; Higgins, J. D.; Taylor, L. S., Impact of surfactants on the crystallization of aqueous suspensions of celecoxib amorphous solid dispersion spray dried particles. Molecular Pharmaceutics 2015,12 (2), 533-541.
Harmon, P.; Galipeau, K.; Xu, W.; Brown, C.; Wuelfing, W. P., Mechanism of dissolution-induced nanoparticle formation from a copovidone-based amorphous solid dispersion. Molecular Pharmaceutics 2016,13 (5), 1467-1481.
Fitriani, L.; Haqi, A.; Zaini, E., Preparation and characterization of solid dispersion freeze-dried efavirenz–polyvinylpyrrolidone K-30. Journal of Advanced Pharmaceutical Technology & Research 2016,7 (3), 105.
Liu, X.; Lu, M.; Guo, Z.; Huang, L.; Feng, X.; Wu, C., Improving the chemical stability of amorphous solid dispersion with cocrystal technique by hot melt extrusion. Pharmaceutical Research 2012,29 (3), 806-817.
Tian, Y.; Jacobs, E.; Jones, D. S.; McCoy, C. P.; Wu, H.; Andrews, G. P., The design and development of high drug loading amorphous solid dispersion for hot-melt extrusion platform. International Journal of Pharmaceutics 2020,586, 119545.
Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B., Amorphous solid dispersions: Rational selection of a manufacturing process. Advanced Drug Delivery Reviews 2016,100, 85-101.
Hancock, B. C.; Shamblin, S. L.; Zografi, G., Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharmaceutical Research 1995,12 (6), 799-806.
Shah, H. S.; Chaturvedi, K.; Dave, R.; Bates, S.; Haware, R. V.; Morris, K. R., New Insights on Warfarin Sodium 2-Propanol Solvate Solid-State Changes Using Multivariate Approach. Crystal Growth & Design 2020.
Shah, H.; Parikh, D.; Butani, S., Formulation development & optimisation of milk dissolving tablets as novel paediatric dosage form. International Journal of Drug Formulation and Reseearch 2014,5, 84-96.
Ivanovska, V.; Rademaker, C. M.; van Dijk, L.; Mantel-Teeuwisse, A. K., Pediatric drug formulations: a review of challenges and progress. Pediatrics 2014,134 (2), 361-372.
Strickley, R. G.; Iwata, Q.; Wu, S.; Dahl, T. C., Pediatric drugs—a review of commercially available oral formulations. Journal of Pharmaceutical Sciences 2008,97 (5), 1731-1774.
Morris, K. R., Structural aspects of hydrates and solvates. Drugs and the Pharmaceutical Sciences 1999,95, 125-182.
Hussain, A. S.; Gurvich, V. J.; Morris, K., Pharmaceutical “new prior knowledge”: twenty-first century assurance of therapeutic equivalence. AAPS PharmSciTech 2019,20 (3), 1-7.