Molecular Structure, Frontier Molecular Orbitals, MESP and UV–Visible Spectroscopy Studies of Ethyl 4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate: A Theoretical and Experimental Appraisal
Vishnu Ashok Adole1 , Bapu Sonu Jagdale1*, Thansing Bhavsing Pawar2, Bhatu Shivaji Desale1
, Bapu Sonu Jagdale1*, Thansing Bhavsing Pawar2, Bhatu Shivaji Desale1
1Department of Chemistry, Arts, Science and Commerce College, Manmad, Taluka-Nandgaon, District- Nashik, India423104. (Affiliated to Savitribai Phule Pune University, Pune (MH), India)
2Research Centre in Chemistry, Loknete Vyankatrao Hiray Arts, Science and Commerce College Panchavati, Nashik-422003, India (Affiliated to Savitribai Phule Pune University, Pune (MH), India)
Corresponding Author Email: jagdalebs@gmail.com
DOI : http://dx.doi.org/10.13005/msri.17.special-issue1.04
Article Publishing History
Article Received on : 09 June 2020
Article Accepted on : 24 July 2020
Article Published : 27 Jul 2020
Plagiarism Check: Yes
Reviewed by: Mr.Sandip S. Pathade
Second Review by: Parashuram Laksminarayana
Final Approval by: Arunabha Roy
Article Metrics
ABSTRACT:
In the current investigation, we wish to report a combined study on the theoretical and experimental investigation of structural, molecular, and spectral properties of ethyl 4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (EDMT). The EDMT molecule is synthesized and characterized by UV-Visible, FT-IR, 1H NMR, 13C NMR, DEPT, and mass spectral techniques. The density functional theory (DFT) investigation was performed by using the B3LYP level of theory at 6-311++G (d,p) basis set. Frontier molecular orbital (FMO) analysis is likewise examined. An TD-DFT method was used for the UV-Visible spectral analysis by using the B3LYP level and 6-311++G (d,p) basis set in the DMSO solvent. Experimental and theoretical UV-Visible spectra were compared in the present study. Various reactivity descriptors are discussed. Besides, Mulliken atomic charges, molecular electrostatic surface potential (MESP), and some valuable thermodynamic functions are studied.
KEYWORDS:
6-311++G (d,p); DFT; Ethyl 4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate; TD-DFT; UV-Visible
Copy the following to cite this article:
Adole V. A, Jagdale B. S, Pawar T. B, Desale B. S. Molecular Structure, Frontier Molecular Orbitals, MESP and UV–Visible Spectroscopy Studies of Ethyl 4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate: A Theoretical and Experimental Appraisal. Mat. Sci. Res. India; Special Issue (2020).
|
Copy the following to cite this URL:
Adole V. A, Jagdale B. S, Pawar T. B, Desale B. S. Molecular Structure, Frontier Molecular Orbitals, MESP and UV–Visible Spectroscopy Studies of Ethyl 4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate: A Theoretical and Experimental Appraisal. Mat. Sci. Res. India; Special Issue (2020). Available from: https://bit.ly/30UmIPs
|
Introduction
3,4-dihydropyrimidin-2(1H)-ones (DHPMs) have developed as useful building blocks for designing new compounds with an expansive assortment of biological applications.1-3 They have gotten a lot of consideration because of the intriguing pharmacological properties related to this heterocyclic framework. DHPMs have been found to display an expansive scope of biological activities such as antitumor,4 antitubercular,5 antimalarial,6 antiviral,7 analgesic,8 antibacterial,9 anti-inflammatory,10 antifungal,10,11 antidiabetic,12 antiproliferative activities,13 etc. Besides, they have additionally been seen as dynamic calcium channel blockers and antihypertensive specialists.14,15 To the extent that the materials science is concerned, the DHMPs have been adequately used to develop heterocyclic compounds with fine optical properties. In materials science, DHPMs are progressively discovering applications in the advancement of materials. To a great extent examined applications in material science are the advancement in functional polymers, adhesives, and fabric dyes.16-18 Eco-friendly approaches are widely accepted and have been used to reduce environmental hazard.19-28 Many green environment-friendly methodologies were researched for creating newer DHPMs as the biological core. The most common synthetic pathways to DHPMs are microwave irradiation, ultrasound irradiation, ionic liquid strategies, nanoparticles as heterogeneous catalysts, grinding, and solvent-free methods.29 DFT computations are reliable and noteworthy for deciding the structural, electronic, and spectral properties of the molecules.30-44 Theoretical quantum computations derived from the DFT strategy have been productively utilized in different fields. The examination of spectroscopic and quantum chemical parameters is by all accounts essential to investigate the chemical behavior of the molecules. Considering all these discussed talked about imperative parts, in this, we wish to report theoretical and experimental studies on molecular structure, HOMO-LUMO analysis, MESP study, and UV–Visible spectral investigation of ethyl 4-(3,4-dimethoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate. In the current research, DFT examination on molecular structure, bond length, bond angle, Mulliken atomic charges have been discussed. The important parameters such as total energy, HOMO-LUMO energies, charge distribution, thermodynamic properties, etc. also studied using the DFT method. Importantly experimental and theoretical comparison on UV–Visible spectra has been discussed.
Methodology
General Remarks
The chemicals with high purity were bought from Local distributor, Nashik. The chemicals were utilized as gotten with no further refinement. NMR spectra were recorded on a sophisticated multinuclear FT NMR Spectrometer model Advance-II (Bruker) with 1H frequency 400 MHz and 13C frequency 100 MHz using DMSO-d6 as a solvent. FT-IR spectra were acquired with potassium bromide pellets on SHIMDAZU spectrometer. Aluminium sheets with silica gel 60 F254 (Merck) were used to monitor the reaction.
Experimental procedure for the synthesis EDMT
A mixture of 3,4-dimethoxy benzaldehyde (0.01 mol), urea (0.01 mol), and ethyl acetoacetate, (0.01 mol) were mixed in 10 mL acetic acid taken in a conical flask. The resulting mixture was stirred on a magnetic stirrer at 70-80 ℃ until the formation of the desired product (checked by TLC). The crude product was transferred into a beaker containing crushed ice, stirred, filtered, dried naturally, and recrystallized to furnished pure white solid (m.p. 178 °C -180 °C) (Scheme 1).
Computational Details
All the calculations were performed utilizing DFT strategy with B3LYP/6-311++G (d,p) basis sets in the Gaussian 03 W program.45 The geometry of the title compound was optimized and the corresponding energy was determined with a 6-311++G (d,p) basis set. Accordingly, the optimized geometrical parameters, energy, atomic charges, dipole moment, and other thermodynamic parameters were calculated theoretically. Also, the theoretical UV-Visible spectrum of the title compound is correlated with the experimental UV-Visible spectrum. Gauss View 4.1 molecular visualization program has been considered to get visual animation. The electronic properties, for example, HOMO–LUMO energies, absorption wavelengths, and oscillator strengths were computed using the B3LYP method of the TD-DFT) and 6-311++G (d,p) basis set.
Results and Discussion
Spectral Analysis
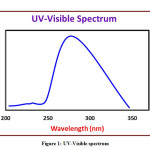
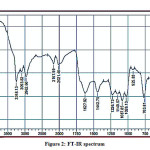
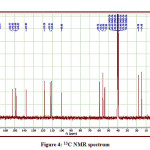
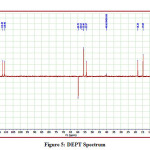
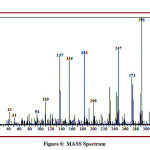
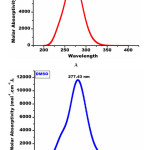
The detailed spectral results are tabulated in Table 1. The UV-Visible spectrum (Figure 1) indicates that the compound EDMT has λmax value 278 nm which corresponds to electronic excitation from HOMO-LUMO transitions. The FT-IR spectrum (Figure 2) confirms various types of stretching and bending vibrations. The vibrational bands at 3248.13 and 3093.82 cm-1 are due to the two N-H groups’ stretching vibrations. The vibrational band at 2962.66 cm-1 is due to sp3 C-H stretching vibrations. The 1627.92 cm-1 absorption signal is because of the carbonyl group flanked between two N-H groups. Other signals are also in acceptable concurrence with the structure of the EDMT molecule. The 1H NMR spectrum (Figure 3) finds the types and the total count of the hydrogen atoms in the molecule. There are total of ten types of protons in the title molecule and therefore has furnished ten signals in the 1H NMR spectrum. The two NH group signals are 9.16 (s, 1H) and 7.69 (s, 1H). All other signals are correctly matched with the structure of the EDMT molecule. The 13C NMR spectrum (Figure 4) predicts types of carbons atoms in the molecule and therefore one can anticipate the skeleton of the molecule. There are total of sixteen types of carbons that have displayed sixteen signals in the 13C NMR spectrum affirming the structure. The two signals 165.94 and 152.77 δ are due to carbonyl carbons of ester and amide groups respectively. From the DEPT spectrum (Figure 5), protonated carbons are differentiated from quaternary carbons. There are eight up signals and one down signal in the DEPT spectrum. Out of the eight up signals, four are methyl and on four are methyne groups. The Mass spectrum (Figure 6) has a molecular ion peak at m/z = 320 and a base peak at m/z = 291.
Table 1: Spectral results of title compound
|
Type of spectra
|
Spectral results
|
|
UV-Visible ( Solvent = DMSO, nm)
|
278.00
|
|
FT-IR (KBr, cm-1)
|
3248.13, 3093.82, 2962.66, 1627.92, 1442.75, 1226.73, 1149.57, 763.81
|
|
1H NMR (400 MHz, DMSO-d6, δ)
|
1.11 (t, J = 7.0 Hz, 3H), 2.24 (s, 3H), 3.71 (s, 6H), 3.99 (q, J = 7.0 Hz, 2H), 5.09 (d, J = 3.8 Hz, 1H), 6.72 (dd, J = 8.2, 2.1 Hz, 1H)), 6.84 (d, J = 2.1 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 7.69 (s, 1H), 9.16 (s, 1H)
|
|
13C NMR (100 MHz, DMSO-d6, δ)
|
14.68, 18.28, 53.96, 55.90, 56.02, 59.70, 99.86, 110.91, 112.21, 118.38, 137.84, 148.53, 148.68, 148.95, 152.77, 165.94
|
|
DEPT (100 MHz, DMSO-d6, δ)
|
14.68 (CH3), 18.28 (CH3), 53.97 (CH), 55.90 (OCH3), 56.02 (OCH3), 110.90 (Ar CH), 112.20 (Ar CH), 118.38 (Ar CH) (all up), 59.70 (OCH2) (down)
|
|
MASS (m/z)
|
320 (M+.), 291, 273, 247, 183, 156, 137, 110
|
Computational Study
Molecular Structure, Bond Lengths and Bond Angle Analysis
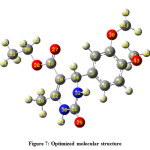
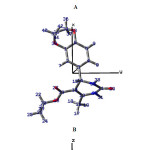
The optimized molecular structure of the title molecule is depicted in Figure 7. Molecular structures A, B, and C (Figure 8) represent presentation along various axes. The optimized molecular geometry provides a good deal of information about the spatial orientation of various atoms in a molecule. From optimized molecular structures, it can be easily seen that the EDMT molecule possesses C1 point group symmetry due to the overall asymmetry of the molecule. Hence, the EDMT molecule is an asymmetric top molecule. This data is a lot of helpful for the determination of various spectroscopic parameters. The structural parameters like bond lengths and bond angles for the title molecule have been established by the DFT/B3LYP method with the 6-311++G (d,p) as basis set and are presented in Table 2. The EDMT molecule comprises of two six-membered rings. The DFT computation predicts the benzene ring is planar (as expected) while the other ring is non-planar. The self-consistent field (SCF) energy of the title molecule at the DFT/B3LYP method with the 6-311++G (d,p) as basis set is found to be −1107.69 a.u. with dipole moment 4.05 Debye. The C10-C12, C13-O27, and C11-O29 bonds are 1.3603, 1.2174, and 1.219 Å long respectively. The bond angles of H21-C20-O28, C2-C1-C6, C1-O38-C39, and H21-C20-O28 are 112.8923 108.6969, 119.8669, 114.9448, and 108.6969° respectively. The bond angle and bond length data are in good agreement with the structure of the title molecule.
Table 2: Optimized geometrical parameters of IPPC by DFT/ B3LYP with 6-311++G (d,p) basis set
|
Bond lengths (Å)
|
|
C1-C2
|
1.3921
|
C11-O29
|
1.219
|
C20-C28
|
1.449
|
|
C1-C6
|
1.4048
|
C11-N30
|
1.402
|
C23-H24
|
1.0926
|
|
C1-O38
|
1.3772
|
C11-N32
|
1.3595
|
C23-H25
|
1.0933
|
|
C2-C3
|
1.3989
|
C12-C16
|
1.5065
|
C23-H26
|
1.0925
|
|
C2-H7
|
1.0835
|
C12-N30
|
1.3855
|
N30-H31
|
1.0088
|
|
C3-C4
|
1.3977
|
C13-O27
|
1.2174
|
N32-H33
|
1.0098
|
|
C3- C14
|
1.5348
|
C13-O28
|
1.3562
|
C34-H35
|
1.0927
|
|
C4- C5
|
1.3931
|
C14-H15
|
1.0905
|
C34-H36
|
1.0897
|
|
C4-H8
|
1.0831
|
C14-N32
|
1.4658
|
C34-H37
|
1.0954
|
|
C5- C6
|
1.392
|
C16-H17
|
1.0918
|
C34-O43
|
1.4349
|
|
C5- H9
|
1.084
|
C16-H18
|
1.088
|
O38-C39
|
1.4345
|
|
C6-O43
|
1.3765
|
C16-H19
|
1.0922
|
C39-H40
|
1.0897
|
|
C10-C12
|
1.3603
|
C20-H21
|
1.0922
|
C39-H44
|
1.0953
|
|
C10-C13
|
1.4673
|
C20-H22
|
1.0921
|
C39-H42
|
1.0931
|
|
C10-C14
|
1.5275
|
C20-C23
|
1.5153
|
–
|
–
|
|
Bond angles (°)
|
|
C2-C1-C6
|
119.8669
|
C10-C12-O30
|
118.535
|
C20- C23-H26
|
111.1729
|
|
C2-C1-H38
|
119.177
|
C16-C12-O30
|
113.8434
|
H24-C23-H25
|
108.1485
|
|
C6-C1-H38
|
120.9221
|
O10-C13-O27
|
123.2155
|
H24-C23-H26
|
108.553
|
|
C1-C2-C3
|
121.0938
|
C10-C13-C28
|
114.9106
|
H25-C23-H26
|
108.1352
|
|
C1-C2-H7
|
118.3975
|
C27-C13-H28
|
121.8701
|
C13-O28-C20
|
115.9493
|
|
C3-C2-H7
|
120.5047
|
C3-C14-N10
|
112.4642
|
C11-N30-N32
|
124.9462
|
|
C2-C3-C4
|
118.6253
|
C3-C14-H15
|
107.5333
|
C11-N30-H31
|
113.4226
|
|
C2-C3-C14
|
119.3121
|
C3-C4-N32
|
112.8923
|
C12-N30-H31
|
119.4272
|
|
C4-C3-C14
|
122.0623
|
H10-C14-N15
|
107.2569
|
C1-N32-C14
|
124.3583
|
|
C3-C4-C5
|
120.5362
|
C10-C14-H32
|
109.1721
|
C11-C12-H33
|
113.9896
|
|
C3-C4-H8
|
120.697
|
C15-C14-H32
|
107.22
|
C14-C12-H33
|
117.9436
|
|
C5-C4-H8
|
118.7648
|
C12-C16-H17
|
110.76
|
H35-C34-H36
|
109.9018
|
|
C4-C5-C6
|
120.7159
|
H12-C16-H18
|
111.2783
|
H35-C34-H37
|
109.7705
|
|
C4-C5-H9
|
120.91
|
H12-C16-H19
|
110.3105
|
H35-C34-O43
|
111.1473
|
|
C6-C5-C9
|
118.3736
|
H17-C16-H18
|
107.0429
|
H36-C34-H37
|
109.4042
|
|
C1-C6-C5
|
119.1268
|
H17-C16-H19
|
108.3867
|
H36-C34-O43
|
106.1786
|
|
C1-C6-C43
|
121.3621
|
H18-C16-H19
|
108.955
|
H37-C34-O43
|
110.3697
|
|
C5-C6-C43
|
119.4421
|
H21-C20-H22
|
107.6967
|
C1-O38-C39
|
114.9448
|
|
C12-C10-C13
|
126.0771
|
H21-C20-C23
|
112.0153
|
O38-C39-H40
|
106.2103
|
|
C12-C10-C14
|
119.3414
|
H21-C20-O28
|
108.6969
|
O38-C39-H41
|
110.4127
|
|
C13-C10-C14
|
114.5592
|
H22-C20-C23
|
112.0176
|
O38-C39-H42
|
111.1475
|
|
O29-C11-N30
|
121.0033
|
H22-C20-O28
|
108.6549
|
H40-C39-H41
|
109.3716
|
|
O29-C11-N32
|
125.3516
|
C23-C20-O28
|
107.6712
|
H40-C39-H42
|
109.868
|
|
N30-C11-N32
|
113.622
|
C20-C23-H24
|
111.189
|
H41-C39-H42
|
109.7619
|
|
C10-C12-C16
|
127.619
|
C20-C23-H25
|
109.5421
|
C6-O43-C34
|
115.0593
|
|
C2-C1-C6
|
119.8669
|
C10-C12-O30
|
118.535
|
C20- C23-H26
|
111.1729
|
|
C2-C1-H38
|
119.177
|
C16-C12-O30
|
113.8434
|
H24-C23-H25
|
108.1485
|
Mulliken Atomic Charges and Molecular Electrostatic Potential Surface Analysis
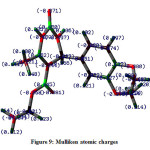
The Mulliken atomic charges depend on the electron density. The charge distribution on the molecules has a crucial role in the field of quantum mechanical calculations for the molecular systems. The pictorial illustration of the Mulliken atomic charges of the EDMT molecule calculated by the DFT/B3LYP method with a 6-311++G (d,p) basis set given in Figure 6 and tabulated in Table 3. Mulliken atomic charges reveal that all the hydrogen atoms have a net positive charge but H31 and H33 hydrogen atoms are highly electropositive with atomic charges of 0.240519 and 0.241399 respectively. This can be ascribed to attachment with a nitrogen atom in both cases. The C11 atom has the highest net positive charge (0.466199) whereas C23 atom has the highest net negative charge (-0.306972) amongst all carbon atoms. Out of the five oxygen atoms, O43 is having more negative charge density of -0.440306 and out of the two nitrogen atoms; N30 is more negative with -0.448179 Mulliken atomic charges.
Table 3: Mulliken atomic charges
|
Atom
|
Charge
|
Atom
|
Charge
|
|
1 C
|
0.104836
|
23 C
|
-0.306972
|
|
2 C
|
-0.003294
|
24 H
|
0.114512
|
|
3 C
|
-0.048593
|
25 H
|
0.113475
|
|
4 C
|
-0.053075
|
26 H
|
0.115685
|
|
5 C
|
-0.068050
|
27 O
|
-0.392430
|
|
6 C
|
0.152106
|
28 O
|
-0.400190
|
|
7 H
|
0.115950
|
29 O
|
-0.389454
|
|
8 H
|
0.100993
|
30 N
|
-0.448179
|
|
9 H
|
0.106250
|
31 H
|
0.240519
|
|
10 C
|
-0.257333
|
32 N
|
-0.404490
|
|
11 C
|
0.466199
|
33 H
|
0.241399
|
|
12 C
|
0.243106
|
34 C
|
-0.104141
|
|
13 C
|
0.455108
|
35 H
|
0.115058
|
|
14 C
|
-0.018689
|
36 H
|
0.122601
|
|
15 H
|
0.162141
|
37 H
|
0.103815
|
|
16 C
|
-0.209945
|
38 O
|
-0.380128
|
|
17 H
|
0.145091
|
39 C
|
-0.104903
|
|
18 H
|
0.148843
|
40 H
|
0.115880
|
|
19 H
|
0.097582
|
41 H
|
0.104293
|
|
20 C
|
-0.008970
|
42 H
|
0.101747
|
|
21 H
|
0.124053
|
43 O
|
-0.440306
|
|
22 H
|
0.127901
|
–
|
–
|
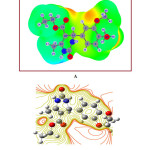
The molecular electrostatic surface potential (MESP) is the three-dimensional portrayal of the charge appropriations on molecules. The MESP diagram plotted by utilizing a 6-311++G (d,p) basis set is represented in Figure 10A and contour plot in Figure 10B. Over the span of on-going years, the MESP has risen as a convincing manual for investigating the molecular interactions. The phenomena like solvent effects, nucleophilic and electrophilic sites, hydrogen bonding forces, etc. could be predicted by the utilization of MESP plots. The different regions of the MESP plot are represented by different colors. The red and yellow surfaces are the regions of large electron density and therefore linked with nucleophilic sites. Similarly, the blue colors indicate low electron density and associated with electrophilic sites. On the other hand, green surfaces suggest regions of zero potential. The MESP proposes, in the EDMT molecule, the benzene ring is highly reactive towards electrophiles. The positive potential is around hydrogen atoms.
Frontier Molecular Orbital and UV-Visible Spectral Analysis
The FMOs, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are extremely crucial to anticipate the reactivity of the molecules. HOMO is the orbital which decides the nucleophilic ability whereas LUMO decides the electrophilic ability of the molecules. The energy gap between these two orbitals helps in deciding the stability. A smaller energy gap indicates high stability. Hence, one can foresee the chemical reactivity of the molecule. The expansion of the HOMO-LUMO energy gap prompts a reduction in adaptability, polarizability, and electron movement in a molecule. The HOMO energy corresponds to ionization enthalpy (I) and LUMO energy to an electron affinity (A). The pictorial outline of the FMOs has been given in Figure 11. The assessment of the FMOs confirms that the electron excitation relates to the transition from the ground state to the first excited state and is essentially depicted by the one-electron move from HOMO to LUMO. In the EDMT molecule, HOMO is seen to be chiefly distributed over the benzene ring and the LUMO is distributed over the enone system. The energy gap between HOMO and LUMO suggests the inevitable charge transfer phenomenon is taking place within the EDMT molecule.
Table 4: Electronic parameters
|
ETotal
(a.u.)
|
EHOMO
(eV)
|
ELUMO
(eV)
|
ΔE
(eV)
|
I
(eV)
|
A
(eV)
|
|
−1107.69
|
−6.39
|
−1.45
|
4.94
|
6.39
|
1.45
|
Note: Abbreviations: I, ionization potential; A, electron affinity; Note: I = −EHOMO & A = −ELUMO
Table 5: Global reactivity parameters
|
χ
(eV)
|
ɳ
(eV)
|
σ
(eV-1)
|
ω
(eV)
|
Pi
(eV)
|
ΔNmax
(eV)
|
Dipole moment (Debye)
|
|
3.92
|
2.47
|
0.40
|
3.11
|
−3.92
|
1.59
|
4.05
|
Note: χ = (I + A)/2; ɳ = (I − A)/2; σ = 1/ɳ; ω = Pi2/2ɳ; Pi = −χ; ΔNmax = −Pi/ɳ. Abbreviations: χ, electronegativity; ɳ, absolute hardness; σ, global softness; ω, global electrophilicity; Pi, chemical potential; ΔNmax, maximum no. of electron transferred.
The different electronic parameters of the EDMT molecule are organized in Table 3 and reactivity descriptors in Table 4. In light of HOMO and LUMO energies and by utilizing Koopmans’ hypothesis different global reactivity descriptors like electronegativity (χ) absolute hardness (ɳ) global softness (σ) global electrophilicity (ω) chemical potential (Pi), the maximum number of electron transferred (ΔNmax) are calculated. The title molecule EDMT is a good electrophile (ω = 3.11 eV). The ΔNmax estimation of the EDMT molecule is 1.59 eV indicating charge transfer. The energy gap is 4.94 eV which indicates an inevitable electron movement within the title molecule. Crucially, the theoretical UV-Visible spectrum of the EDMT is correlated with the experimental UV-Visible spectrum to aid the computed results. The theoretical UV-Visible absorption peaks are 283.51 nm in the gas phase and 277.43 nm (Figure 12A) in the DMSO solvent (Figure 12B). The experimental UV-Visible absorption peak is 278.00 nm in the DMSO solvent. The great agreement between the theoretical and experimental UV-Visible absorption values supports the computed results. Absorption energies (λ in nm), excitation energies, electronic transitions, and oscillator strength (ƒ) of EDMT are computed at TD-B3LYP/6-311++G (d,p) level of theory for B3LYP/6-311++G (d,p) optimized geometry in the gas phase and DMSO solvent. The absorption band is slightly shifted to a lower wavelength from the gas phase to the DMSO solvent. At the point when the ground state is more polar than the excited state, the polar solvents settle the ground state more than the excited state. So in general there is an expansion in the energy gap between the ground state and excited state bringing about the blue shift.
Table 6: Absorption energies (λ in nm), Oscillator strength (ƒ), and transitions of EDMT computed at TD-B3LYP/6-311G++ (d,p) level of theory for B3LYP/6-311G ++ (d,p) optimized geometry in gas phase
|
Sr. No.
|
Absorption
Wavelength
(nm)
|
Excitation energy
(eV)
|
Excited
state
|
Electronic
Transition
|
Oscillator strength
(f)
|
|
1
|
283.51
|
4.3732
|
1
|
85 -> 86
|
0.62706
|
|
2
|
269.94
|
4.5930
|
2
|
84 -> 86
|
0.62706
|
|
3
|
261.36
|
4.5930
|
3
|
83 -> 86
|
0.66620
|
|
4
|
245.66
|
5.0470
|
4
|
83 -> 87
|
0.10131
|
Table 7: Absorption energies (λ in nm), Oscillator strength (ƒ), and transitions of EDMT computed at TD-B3LYP/6-311G++ (d,p) level of theory for B3LYP/6-311G ++ (d,p) optimized geometry in DMSO solvent
|
Sr. No.
|
Absorption
Wavelength
(nm)
|
Excitation energy
(eV)
|
Excited
state
|
Electronic
Transition
|
Oscillator strength
(f)
|
|
1
|
277.43
|
4.4691
|
1
|
85 -> 86
|
0.68511
|
|
2
|
273.97
|
4.5255
|
2
|
84 -> 86
|
0.67699
|
|
3
|
254.77
|
4.8665
|
3
|
83 -> 86
|
0.62180
|
|
4
|
244.66
|
5.0675
|
4
|
83 -> 87
|
0.24938
|
Thermodynamic Parameter Analysis
Considering the vibrational assessment, the distinctive thermodynamic properties were presented from the computed vibrational frequencies and are recorded in Table 6. All the thermodynamic data revealed in this would be valuable for additional assessment and could be used to infer other thermodynamic boundaries as demonstrated by connections of thermodynamic capacities.
Table 8: Thermochemical information of EDMT
|
Parameter
|
Value
|
|
E total (kcal mol-1)
Translational
Rotational
Vibrational
|
228.433 0.889
0.889
226.656
|
|
Heat Capacity at constant volume,
Cv (cal mol-1K-1)
Translational
Rotational
Vibrational
|
78.060
2.981
2.981
72.098
|
|
Total entropy S
(cal mol-1K-1)
Translational
Rotational
Vibrational
|
144.603
43.186
35.147
66.270
|
|
Zero point Vibrational Energy Ev0 (Kcal mol-1)
|
216.07087
|
|
Rotational constants (GHZ)
|
0.30045 0.16655 0.13128
|
Conclusions
In outline, the EDMT molecule is synthesized by a three-component reaction. The structure of the EDMT molecule is affirmed based on the basis of UV-Visible, FT-IR, 1H NMR, 13C NMR, and Mass spectral methods. The geometry of the title molecule was optimized by using a 6-311++G (d,p) basis set, and the geometrical parameters like bond lengths and bond angles were computed at the same level of theory. The title molecule is an asymmetric top molecule with C1 point group symmetry. The FMO study is discussed and various chemical, electronic, and quantum chemical parameters are studied to analyze the chemical reactivity of the title molecule. The HOMO-LUMO energy gap suggests that the charge transfer phenomenon is taking place within the molecule. The theoretical wavelength in the gas phase is higher than in the DMSO solvent. The theoretical UV-Visible spectrum is correlated with the experimental UV-Visible spectrum and a very nice agreement is found. The MESP plot reveals that electrophiles would attack at the benzene ring. The thermodynamic properties like total energy, total molar heat capacity, total entropy, zero-point vibrational energy, and rotational constants have been figured and discussed.
Acknowledgments
Authors acknowledge central instrumentation facility (CIF), Savitribai Phule Pune University, Pune for NMR, MGV’s Pharmacy College, Nashik for UV-Visible, and central instrumentation centre (CIC), KTHM College, Nashik for FT-IR spectral analysis. Authors also would like to thank Lokenete Vyankatrao Hiray Arts, Science and Commerce College Panchavati, Nashik and Arts, Science and Commerce College, Manmad for permission and providing necessary research facilities. Authors would like to express their sincere and humble gratitude to Prof. (Dr.) Arun B. Sawant for Gaussian study. Dr. Aapoorva P. Hiray, Coordinator, MG Vidyamandir institute, is gratefully acknowledged for the Gaussian package.
Funding Soure
We have not received any kind of fund for the research work.
Conflict of Interest
Authors declared that he do not have any conflict of interest regarding this research article.
References
- de Fatima, A., Braga, T.C., Neto, L.D.S., Terra, B.S., Oliveira, B.G., da Silva, D.L. and Modolo, L.V., 2015. A mini-review on Biginelli adducts with notable pharmacological properties. Journal of advanced research, 6(3), pp.363-373.
- Matos, L.H.S., Masson, F.T., Simeoni, L.A. and Homem-de-Mello, M., 2018. Biological activity of dihydropyrimidinone (DHPM) derivatives: A systematic review. European journal of medicinal chemistry, 143, pp.1779-1789.
- Kaur, R., Chaudhary, S., Kumar, K., Gupta, M.K. and Rawal, R.K., 2017. Recent synthetic and medicinal perspectives of dihydropyrimidinones: A review. European journal of medicinal chemistry, 132, pp.108-134.
- Mostafa, A.S. and Selim, K.B., 2018. Synthesis and anticancer activity of new dihydropyrimidinone derivatives. European journal of medicinal chemistry, 156, pp.304-315.
- Singh, K., Singh, K., Wan, B., Franzblau, S., Chibale, K. and Balzarini, J., 2011. Facile transformation of Biginelli pyrimidin-2 (1H)-ones to pyrimidines. In vitro evaluation as inhibitors of Mycobacterium tuberculosis and modulators of cytostatic activity. European journal of medicinal chemistry, 46(6), pp.2290-2294.
- October, N., Watermeyer, N.D., Yardley, V., Egan, T.J., Ncokazi, K. and Chibale, K., 2008. Reversed Chloroquines Based on the 3, 4‐Dihydropyrimidin‐2 (1H)‐one Scaffold: Synthesis and Evaluation for Antimalarial, β‐Haematin Inhibition, and Cytotoxic Activity. ChemMedChem: Chemistry Enabling Drug Discovery, 3(11), pp.1649-1653.
- Kumarasamy, D., Roy, B.G., Rocha-Pereira, J., Neyts, J., Nanjappan, S., Maity, S., Mookerjee, M. and Naesens, L., 2017. Synthesis and in vitro antiviral evaluation of 4-substituted 3, 4-dihydropyrimidinones. Bioorganic & medicinal chemistry letters, 27(2), pp.139-142.
- Ajitha, M., Rajnarayana, K. and Sarangapani, M., 2011. Synthesis and evaluation of new 3-substituted-[3, 4-dihydropyrimidinones]-indolin-2-ones for analgesic activity. International Res J Pharm, 2(7), pp.80-84.
- Attri, P., Bhatia, R., Gaur, J., Arora, B., Gupta, A., Kumar, N. and Choi, E.H., 2017. Triethylammonium acetate ionic liquid assisted one-pot synthesis of dihydropyrimidinones and evaluation of their antioxidant and antibacterial activities. Arabian Journal of chemistry, 10(2), pp.206-214.
- Tale, R.H., Rodge, A.H., Hatnapure, G.D. and Keche, A.P., 2011. The novel 3, 4-dihydropyrimidin-2 (1H)-one urea derivatives of N-aryl urea: synthesis, anti-inflammatory, antibacterial and antifungal activity evaluation. Bioorganic & medicinal chemistry letters, 21(15), pp.4648-4651.
- Singh, O.M., Singh, S.J., Devi, M.B., Devi, L.N., Singh, N.I. and Lee, S.G., 2008. Synthesis and in vitro evaluation of the antifungal activities of dihydropyrimidinones. Bioorganic & medicinal chemistry letters, 18(24), pp.6462-6467.
- Dhumaskar, K.L., Meena, S.N., Ghadi, S.C. and Tilve, S.G., 2014. Graphite catalyzed solvent free synthesis of dihydropyrimidin-2 (1H)-ones/thiones and their antidiabetic activity. Bioorganic & medicinal chemistry letters, 24(13), pp.2897-2899.
- Russowsky, D., Canto, R.F., Sanches, S.A., D’Oca, M.G., de Fátima, Â., Pilli, R.A., Kohn, L.K., Antoˆnio, M.A. and de Carvalho, J.E., 2006. Synthesis and differential antiproliferative activity of Biginelli compounds against cancer cell lines: monastrol, oxo-monastrol and oxygenated analogues. Bioorganic chemistry, 34(4), pp.173-182.
- Singh, K., Arora, D., Singh, K. and Singh, S., 2009. Genesis of dihydropyrimidinonep calcium channel blockers: recent progress in structure-activity relationships and other effects. Mini reviews in medicinal chemistry, 9(1), p.95.
- Rovnyak, G.C., Atwal, K.S., Hedberg, A., Kimball, S.D., Moreland, S., Gougoutas, J.Z., O’Reilly, B.C., Schwartz, J. and Malley, M.F., 1992. Dihydropyrimidine calcium channel blockers. 4. Basic 3-substituted-4-aryl-1, 4-dihydropyrimidine-5-carboxylic acid esters. Potent antihypertensive agents. Journal of medicinal chemistry, 35(17), pp.3254-3263.
- Boukis, A.C., Llevot, A. and Meier, M.A., 2016. High glass transition temperature renewable polymers via Biginelli multicomponent polymerization. Macromolecular rapid communications, 37(7), pp.643-649.
- Zhao, Y., Yu, Y., Zhang, Y., Wang, X., Yang, B., Zhang, Y., Zhang, Q., Fu, C., Wei, Y. and Tao, L., 2015. From drug to adhesive: a new application of poly (dihydropyrimidin-2 (1 H)-one) s via the Biginelli polycondensation. Polymer Chemistry, 6(27), pp.4940-4945.
- Patil, S.R., Choudhary, A.S., Patil, V.S. and Sekar, N., 2015. Synthesis, optical properties, dyeing study of dihydropyrimidones (DHPMs) skeleton: Green and regioselectivity of novel Biginelli scaffold from lawsone. Fibers and Polymers, 16(11), pp.2349-2358.
- Adole, V.A., Jagdale, B.S., Pawar, T.B. and Sagane, A.A., 2020. Ultrasound promoted stereoselective synthesis of 2,3-dihydrobenzofuran appended chalcones at ambient temperature. South African Journal of Chemistry, 73, pp.35-43.
- Adole, V.A., Pawar, T.B., Koli, P.B. and Jagdale, B.S., 2019. Exploration of catalytic performance of nano-La 2 O 3 as an efficient catalyst for dihydropyrimidinone/thione synthesis and gas sensing. Journal of Nanostructure in Chemistry, 9(1), pp.61-76.
- Adole, V.A., Pawar, T.B. and Jagdale, B.S., 2019. Aqua‐mediated rapid and benign synthesis of 1, 2, 6, 7‐tetrahydro‐8H‐indeno [5,4‐b] furan‐8‐one‐appended novel 2‐arylidene indanones of pharmacological interest at ambient temperature. Journal of the Chinese Chemical Society.67:306-315.
- Narayanan, D.P., Gopalakrishnan, A., Yaakob, Z., Sugunan, S. and Narayanan, B.N., 2020. A facile synthesis of clay–graphene oxide nanocomposite catalysts for solvent free multicomponent Biginelli reaction. Arabian Journal of Chemistry, 13(1), pp.318-334.
- Adole, V.A., More, R.A., Jagdale, B.S., Pawar, T.B. and Chobe, S.S., 2020. Efficient Synthesis, Antibacterial, Antifungal, Antioxidant and Cytotoxicity Study of 2‐(2‐Hydrazineyl) thiazole Derivatives. ChemistrySelect, 5(9), pp.2778-2786.
- Kadam, V.V., Waghchaure, R.H. and Pawar, T.B., 2020. Green Synthetic Approaches in Organic Synthesis: A Short Review. 8(4), 12-15.
- Kazemi, M., 2020. Magnetically reusable nanocatalysts in biginelli synthesis of dihydropyrimidinones (DHPMs). Synthetic Communications, 50(10), pp.1409-1445.
- Adole, V.A., Synthetic approaches for the synthesis of dihydropyrimidinones/ thiones (biginelli adducts): a concise review. World journal of pharmaceutical research, 9(6), 1067-1091.
- Khemalapure, S.S., Hiremath, S.M., Hiremath, C.S., Katti, V.S. and Basanagouda, M., 2020. Investigations of Structural, Vibrational and electronic properties on 5-(6-methyl-benzofuran-3-ylmethyl)-3H-[1, 3, 4] oxadiazole-2-thione: Experimental and Computational Approach. Chemical Data Collections, p.100410.
- Adole, V.A., Waghchaure, R.H., Jagdale, B.S. and Pawar, T.B., 2020. Investigation of Structural and Spectroscopic Parameters of Ethyl 4-(4-isopropylphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate: a DFT Study. Chemistry & Biology Interface, 10(1), 22-20.
- Balakrishnan, A., Sambanthan, M., Murugesan, R. and Sundaram, S., 2020. Molecular structure, spectroscopic (FT-IR, FT-Raman, NMR, UV-VIS), chemical reactivity and biological examinations of Ketorolac. Journal of Molecular Structure, p.128040.
- Adole, V.A., Jagdale, B.S., Pawar, T.B. and Sawant, A.B., 2020. Experimental and theoretical exploration on single crystal, structural, and quantum chemical parameters of (E)‐7‐(arylidene)‐1,2,6,7‐tetrahydro‐8H‐indeno [5,4‐b] furan‐8‐one derivatives: A comparative study. Journal of the Chinese Chemical Society. https://doi.org/10.1002/jccs.202000006.
- Hiremath, S.M., Khemalapure, S.S., Hiremath, C.S., Patil, A.S. and Basanagouda, M., 2020. Quantum chemical computational and spectroscopic (IR, Raman, NMR, and UV) studies on the 5-(5-methoxy-benzofuran-3-ylmethyl)-3H-[1, 3, 4] oxadiazole-2-thione. Journal of Molecular Structure, p.128041.
- Waghchaure, R.H. and Pawar, T.B., 2020. SYNTHESIS, CHARACTERIZATION, MOLECULAR STRUCTURE, AND HOMO-LUMO STUDY OF 2-PHENYLQUINOXALINE: A DFT EXPLORATION. World journal of pharmaceutical research, 9(6), 1867-1881.
- Tankov, I. and Yankova, R., 2019. Hirshfeld surface, DFT vibrational (FT-IR) and electronic (UV–vis) studies on 4-amino-1H-1, 2, 4-triazolium nitrate. Journal of Molecular Structure, 1179, pp.581-592.
- Adole, V.A., Waghchaure, R.H., Jagdale, B.S., Pawar, T.B. and Pathade, S.S., Molecular tructure, frontier molecular orbital and spectroscopic examination on dihydropyrimidinones: a comparative computational approach Journal of Advanced Scientific Research, 2020. 11 (2), pp.64-70.
- Pawar, T.B., Jagdale, B.S., Sawant, A.B. and Adole, V.A., 2017. DFT Studies of 2-[(2-substitutedphenyl) carbamoyl] benzoic acids. Journal of Chemical, Biological and Physical Sciences, 7, pp.167-175.
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery Jr, J.A., Vreven, T., Kudin, K.N., Burant, J.C. and Millam, J.M., Gaussian 03 (Revision E. 01), Gaussian, Inc, Wallingford CT, 2004.
The optimized molecular structure of the title molecule is depicted in Figure 7. Molecular structures A, B, and C (Figure 8) represent presentation along various axes. The optimized molecular geometry provides a good deal of information about the spatial orientation of various atoms in a molecule. From optimized molecular structures, it can be easily seen that the EDMT molecule possesses C1 point group symmetry due to the overall asymmetry of the molecule. Hence, the EDMT molecule is an asymmetric top molecule. This data is a lot of helpful for the determination of various spectroscopic parameters. The geometrical parameters like bond lengths and bond angles for the title molecule have been set up by the DFT/B3LYP strategy with the 6-311++G (d,p) as premise set and are introduced in Table 2. The EDMT molecule comprises of two six-membered rings. The DFT computation predicts the benzene ring is planar (as expected) while the other ring is non-planar. The self-consistent field (SCF) energy of the title molecule at the DFT/B3LYP method with the 6-311++G (d,p) as basis set is found to be −1107.69 a.u. with dipole moment 4.05 Debye. The C10-C12, C13-O27, and C11-O29 bonds are 1.3603, 1.2174, and 1.219 Å long respectively. The bond angles of H21-C20-O28, C2-C1-C6, C1-O38-C39, and H21-C20-O28 are 112.8923 108.6969, 119.8669, 114.9448, and 108.6969° respectively.The bond angle and bond length data are in good agreement with the structure of the title molecule.

This work is licensed under a Creative Commons Attribution 4.0 International License.