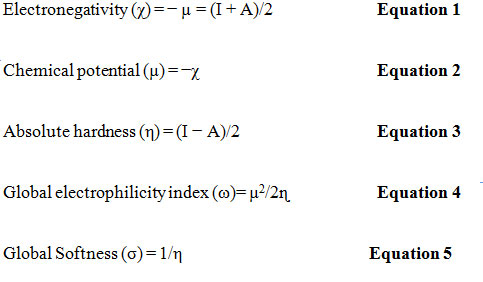Synthesis, Molecular Structure, HOMO-LUMO, Chemical, Spectroscopic (UV-Vis and IR), Thermochemical Study of Ethyl 6-amino-5-cyano-2-methyl-4-(4-nitrophenyl)-4H-pyran-3-carboxylate: A DFT Exploration
Vishnu A. Adole1*, Tejendra A. Rajput2 and Bapu S. Jagdale3
1Department of Chemistry, Mahatma Gandhi Vidyamandir’s Arts, Science and Commerce College (Affiliated to Savitribai Phule Pune University, Pune) Manmad, Nashik, MS, India.
2Department of Chemistry, Arts, Commerce and Science College (Affiliated to Mumbai University, Mumbai) Onde, Palghar, MS, India.
3Department of Chemistry, Mahatma Gandhi Vidyamandir’s Loknete Vyankatrao Hiray Arts, Science and Commerce College (Affiliated to Savitribai Phule Pune University, Pune) Panchavati, Nashik, MS, India.
Article Publishing History Article Received on : 11-Jul-2021 Article Accepted on : 02-Sep-2021 Article Published : 04 Sep 2021 Plagiarism Check: Yes Reviewed by: Dr. Sandip Pathade Second Review by: Dr. Thansing Pawar Final Approval by: Dr. Saleheh Abbaspoor
Article Metrics
ABSTRACT:
The ethyl 6-amino-5-cyano-2-methyl-4-(4-nitrophenyl)-4H-pyran-3-carboxylate (ACNPPC) was synthesized using an environmentally friendly method and looked into in terms ofstructural, UV-visible, vibrational, and computational analysis. In the gaseous phase, calculations of the density functional theory (DFT) with B3LYP/6-311G(d,p) level were performed. Using Time-dependent density functional theory (TD-DFT) with the B3LYP/6-311G(d,p) basis set method, the HOMO and LUMO energies are calculated. For assessing electrophilic and nucleophilic reactive sites, the molecular electrostatic surface potential (MESP) and contour plot were plotted over the optimized structure. Using computed and experimental vibrational spectra, vibrational assignments were elucidated. To illustrate the charge density in the title compound, Mulliken atomic charges are disclosed. In addition, using vibrational analysis, some thermochemical functions have also been derived. Theoretical simulations have shown the best relationship with experimental results obtained with the B3LYP/6-311G(d,p) level of theory at the DFT and TD-DFT methods.
Adole V. A, Rajput T. R, Jagdale B. S. Synthesis, Molecular Structure, HOMO-LUMO, Chemical, Spectroscopic (UV-Vis and IR), Thermochemical Study of Ethyl 6-amino-5-cyano-2-methyl-4-(4-nitrophenyl)-4H-pyran-3-carboxylate: A DFT Exploration. Mat. Sci. Res. India;18(2).
Copy the following to cite this URL:
Adole V. A, Rajput T. R, Jagdale B. S. Synthesis, Molecular Structure, HOMO-LUMO, Chemical, Spectroscopic (UV-Vis and IR), Thermochemical Study of Ethyl 6-amino-5-cyano-2-methyl-4-(4-nitrophenyl)-4H-pyran-3-carboxylate: A DFT Exploration. Mat. Sci. Res. India;18(2). Available from: https://bit.ly/2WOdRRE
Introduction
The pyran is a six-membered heterocyclic, non-aromatic ring made of five carbon atoms and one oxygen atom with two double bonds.The saturated carbon in 2H-pyran is at position 2, while the saturated carbon in 4H-pyran is at the C4 position. Pyran derivatives are a valuable class of heterocyclic compounds commonly found in nature.1In a variety of natural products, the fused pyran ring skeleton is a well-known heterocycle and an essential component. Owing to their interaction with different kinds of biological properties, pyran derivatives have attracted a great deal of interest.2,3In the modern era, researchers from both academia and industry have drawn significant interest in the pyran scaffold. Pyran scaffold-containing heterocyclic compounds are of great interest because they have a wide spectrum of biological properties. These include anticancer4,analgesic5, cytotoxic6, anti-tumour7, anti-HIV8, antimicrobial9,10, etc. A number of green methods have been reported to synthesize variety of heterocyclic compounds11-15. Likewise, pyran derivatives have also been synthesized by variety of green chemistry routes such as Amberlyst A21, N-methylimidazole, Magnetic AluminosilicateNanoclay, iodine, visible light-induced, etc16-20. Here in the current research, we have used catalytic amount of sulfamic acid under stirring condition at 80 ℃ in PEG-400-H2O (8:2)reaction mediato drive the reaction to afford the pure product.
For the title compound, the literature review showed that no comprehensive quantum chemical analysis was carried out. This research was therefore carried out and the spectroscopic characterization of ACNPPC by means of UV, IR, 1H NMR spectra has been reported in this paper. DFT computations provide good deal of structural and spectroscopic information by using suitable basis set21-32. The experimental findings were wellsupported by the quantum chemical simulations. This has been accomplished using the method of density functional theory (B3LYP) with a 6-311G(d,p) basis set. The title compound was assessed for molecular and structural properties such as dipole moment, molecular electrostatic potential, thermodynamic parameters, and bond lengths and bond angles. The frontier molecular orbitals such as HOMO and LUMO clearly state how the molecular interactions occur, so that the chemical reactivity of the molecules can be defined. With the support of simulated electronic and vibrational spectra; UV-Visible and Vibrational assignments were made. To the best of our knowledge, this is the primary research on DFT investigation of the title compound.
Experimental
Details
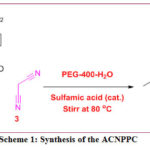
Synthesis
of the title compound
In a typical synthesis procedure (Scheme 1), ethylacetoacetate (1, 0.01 mol) and 4-nitrobenzaldehyde (2, 0.01 mol) were mixed initially in 15 mL PEG-400-H2O (8:2).This reaction mixture was stirred at 80 ℃ until dissolution of 4-nitrobenzaldehyde. Then, catalytic amount of sulfamic acid was added and the reaction mixture was stirred at 80 ℃ for half an hour. After this malononitrile(3, 0.01 mol) was added slowly and reaction was continued further until the formation of the pure product (4). Reaction was continuously monitored by using thin layer chromatography. The crude product was obtained after adding reaction mass into the ice-cold water and then filtered and dried. The crude product was then recrystallized to afford yellow crystals of pure product.
DFT simulations were performed using the Gaussian-03 programme package on an Intel (R) Core (TM) i5 computer with no geometry constraints33. Molecular structure was optimized using DFT method with B3LYP (Becke three-parameter Lee–Yang–Parr) exchange-correlation functional34, 35. The 6-311G(d,p) basis set was used to optimize the molecular structure. The same basis set was employed on the fully DFT optimized geometries to predict the harmonic vibrational frequencies. The Gauss View 4.1.2 molecular visualization Programme was used to establish optimized geometry, FMO, MESP and contour plots36. The Time-dependent density-functional theory (TD-DFT) with 6-311G(d,p) basis set was used to predict the vertical excitation energy and electronic absorption spectra. Both in gas phase and in DMSO, the simulated electronic absorption spectra were computed. TD-DFT computations were also used to examine electronic configurations, oscillator strengths, and excited state energies.Ionization potential, electron affinity, electronegativity, chemical hardness, global softness, global electrophilicity, and chemical potential were derived using HOMO and LUMO energy values.The thermodynamic parameters of the title compound were also determined, demonstrating the relationship between heat capacity at constant volume (Cv) and entropy (S).
Results
and Discussion
Chemistry
The titled compound was synthesized with excellent yield by reacting ethylacetoacetate, nitrobenzaldehyde and malononitrile. PEG-400-H2O solvent system in presence of catalytic amount of sulfamic acid is driving the reaction to completion within short period of time. PEG-400-H2O was revealed to be a cost-effective, non-toxic, and efficient medium for the one-pot synthesis of the titled compound with high yield. This solvent system is notable for its environmental friendliness, low cost, high yields, and recyclability. The reaction product was characterized by spectral methods like IR, 1H NMR, and 13C NMR. Thetheoretical absorption maxima wavelength in UV spectrum of the titled compound was show up at 343.94 nm in the DMSO unmistakably demonstrated a hypsochromic shift is occurring in the DMSO. The IR spectra of titled compoundshowed the appearance of the broad intense peaks in the range of 3407-3338cm-1due to presence of N-H stretching vibrations. The peak at2985.45 indicate presence of –C-H stretching vibrations, and the intense peak at 2202.88 cm-1 clearly shows presence of nitrile group in the compound. The νCO stretching vibrations of ester function are incredibly observed at 1682.30 cm-1.The presence of nitro group in the compound is affirmed by FT-IR by observing two important vibrations at 1532 cm-1 and 1356 cm-1 due to asymmetric and symmetric vibrations respectively. The existence of ethyl (CH3-CH2-) groups attached to oxygen atom was confirmed by the signals 4.01 (q, 2 H) and 1.19 (t, 3H) in the 1H NMR. Two Signals correspond to four de-shielded hydrogen atoms in the aromatic region (8.01 ppm d, 2 H and 7.54 ppm d, 2 H) ppm in the 1H NMR spectrademonstrate presence of nitro group on the benzene. Moreover, the signal which appeared as broad singlet at 6.89 ppm indicates exchangeable proton on nitrogen, also singlet at 2.40 ppm correspond to three hydrogens confirms methyl group attached to sp2 hybridized carbon of carbon-carbon double bond. In the 13C NMR spectrum, fourteen signals are appeared that confirms the carbon skeleton of the titled compound.
Computational
study
Molecular geometry
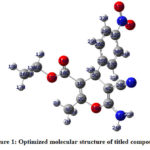
The geometrical parameters; bond lengths and bond angles of the title compound ACNPPClisted in Table 1and Table 2 respectively were determined using DFT/B3LYP method with 6-311G(d,p) basis set. The optimized molecular structure of the title compound obtained from Gaussian 03W and Gauss View 4.1.1 programme are shown in Figure 1. The C-H bond length varied from 1.0809 Å to 1.0945 Å. The C-C bond lengths are found in the range of 1.3466 Å to 1.5339 Å. The C-N bond length of amino group is found be 1.362 Å. On the contrary, C-N bond length of nitro group is 1.4779 Å. The greater C-N bond length of nitro group as compared to the amino group is due the fact that the nitro group is involved in strong resonance effect with the aromatic ring. The N=O bond of the nitro group are found to have nearly same bond length. The carbonyl group is having 1.2116 Å. Out of three C-O bonds, C2-O38 is the longest with the bond length value of 1.3882 Å. The nitrile group has bond length value of 1.1591 Å.
Figure 1: Optimized molecular structure of titled compound.
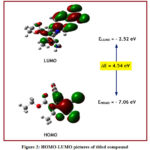
One of the most important aspects employed in quantum chemistry is the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).As an electron donor, HOMO, which is known as the outermost orbital containing electrons, appears to give away these electrons. LUMO, on the other hand, is the innermost orbital comprising electron acceptance areas.HOMO energy is straightforwardly identified with ionization potential, and LUMO energy is straightforwardly identified with electron affinity. Figure 2 demonstrates the pictorial image of HOMO and LUMO.The energy for HOMO is –7.06 eV and the energy for LUMO is –2.52 eV. The energy gap that serves as an important stability factor for the compound is called the energy difference between the HOMO and LUMO orbitals. The HOMO-LUMO energy gap for the title compound is 4.54 eV, confirming that the structure of the molecule is stable. Lower the energy gap; electrons are excited more readily from the ground state to excited state.Global quantum chemical identifiers such as electronegativity (χ), chemical potential (μ), global hardness (η), global softness (σ) and global electrophilicity index can be described by the use of HOMO and LUMO energy values. The formulae to calculate global reactivity descriptors are as given below-
Where, I = ionization potential = − EHOMOandA = electron affinity = − ELUMO
The quantities of global quantum chemical identifiers are included in Table 3.The ionization potential value revealed that to extract an electron from the HOMO, energy of 7.06 eV is required. The lower electron affinity value (2.54 eV) demonstrates more prominent chemical reactivity with the nucleophiles. The greater molecular hardness (2.27 eV) associated with the molecule is evidenced by higher hardness and lower softness values (0.45eV-1).The electrophilicity index (5.07 eV) aids in the description of the title compound’s activity.
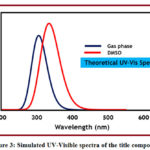
The theoretical absorption spectra for the optimised compounds in the gas phase and DMSO were estimated using the TD-DFT method with the B3LYP/6-311G(d,p) basis set in the gas phase and DMSO.The simulated UV-Visible spectra are given in Figure 3.Table 4 shows the excited states that were taken into account for the calculation. In the gas phase, the absorption maxima wavelength is 357.70 nm, while in DMSO, it is 343.94 nm. This evidently showed that the DMSO is undergoing a blue shift.This absorption corresponds to three transitions in the gas phase and two transitions in the DMSO. Likewise second excited state is also composed of same number of transitions in the gas phase and DMSO. The second, third and fourth gas phase excited states are found to be situated at 349.04, 305.80, and 305.13 nm respectively. However, in the DMSO they were found at 342.20, 329.55, and 327.07 nm. This absorption data suggests blue shift for first two excited states and red shift for the third and fourth excited states when solvent is shifted to DMSO from the gas phase.The blue shift for the first two excited states is attributed to the stabilization of ground state in DMSO solvent. However, for the third and fourth excited states excited state is more stabilized thereby causing red shift. The third and fourth excited states in the gas phase comprises four and two transitions and in the DMSO, four and six. In conclusion, DMSO was found to increase the HOMO-LUMO energy gap thereby causing hypsochromic shift.
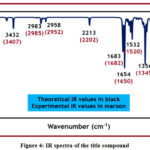
Figure 3: Simulated UV-Visible spectra of the title compound.
Table 4: Electronic absorption data obtained using the TD-DFT method at B3LYP/6-311G(d,p) basis set
State
Gas Phase
DMSO
Configuration
f
Wavelength
(nm)
Configuration
Wavelength
(nm)
f
1
83 -> 87
83 -> 88
83 -> 91
0.0003
357.70
82 -> 87
82 -> 91
343.94
0.0169
2
79 -> 87
86 -> 87
86 -> 88
0.0002
349.04
82 -> 87
82 -> 87
342.20
0.0249
3
84 -> 87
85 -> 87
85 -> 88
86 -> 87
86 -> 88
0.0307
305.80
84 -> 87
85 -> 87
85 -> 88
86 -> 88
329.55
0.0516
4
82 -> 87
82 -> 91
0.0634
305.13
80 -> 88
81 -> 88
84 -> 87
85 -> 87
85 -> 88
86 -> 88
327.07
0.0205
Vibrational and
thermochemical study
Figure 4 shows the IR spectra of the title compound. Scaled IR values and experimental IR values are displayed in various colours in the accompanying spectra. The vibrational bands were correctly assigned owing to a comparison of experimental and scaled IR measurements.The theoretically expected N-H vibrational stretching is found at 3432 cm-1 in the IR spectrum and 3407 cm-1 in the experimental one. This indicates that the two compared values are very much in agreement. The experimental values for similar vibrations are 2985 cm-1 and 2952 cm-1, whereas the cm-1 aliphatic C-H stretching vibrations are observed at 2983 cm-1 and 2958 cm-1. In the predicted and actual IR spectra, nitrile stretching is seen at 2213 cm-1 and 2202 cm-1, respectively.The C=C stretching vibrations are also greatly matching and found at 1654 cm-1 and 1650 cm-1 in the theoretical and experimental IR spectra respectively. The another two important vibrations at 1532 cm-1 and 1356 cm-1 in the experimental IR spectrum are assigned to the asymmetric and symmetric vibrations of the nitro group by comparing with scaled vibrational wavenumbers which are found at 1520 cm-1 and 1345 cm-1. In this method, the comparison has been used to adequately assign vibrational bands to the title compounds, and the compared IR spectra have a very good relationship.
Table 5 lists the typical thermodynamic functions for the title compound, such as total thermal energy, heat capacity (Cv), and entropy (S), which were calculated using vibrational analysis.This data serves as a useful guide on the title compound for further analysis. This can be used for other thermodynamic energies to be computed.
Table 5: Thermochemical properties of the title compound
Thermochemical property
Value
Total thermal energy(E, KCal/Mol)
199.803
0.889 (Translational)
0.889 (Rotational)
198.025 (Vibrational)
Total molar heat capacity (CV,Cal/Mol-Kelvin)
83.677
2.981 (Translational)
2.981 (Rotational)
77.715 (Vibrational)
Total molar entropy(S, Cal/Mol-Kelvin)
167.231
43.269 (Translational)
43.269 (Rotational)
88.659 (Vibrational)
Molecular Electrostatic surface Potential and
Mulliken atomic charges
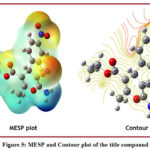
Molecular electrostatic surface potential is a three-dimensional picture that can be used to represent the distributions of charge and charge-related properties. It demonstrates the probable positions for electrophilic attacks and is useful in processes of biological comprehension and interactions of hydrogen bonding.Different color shades show the electrostatic potential on the compound’s surface. The red colour denotes locations with the highest electronegative electrostatic potential, whereas the blue colour denotes areas with the highest positive electrostatic potential, and the green colour denotes areas with no electrostatic potential.The MESP and Contour plot of the title compound (Figure 5) at the DFT level is plotted using the molecular visualization program Gauss View 4.1.2. The attraction of the proton by the electron density (shades of red and yellow) is the negative electrostatic potential, whereas the repulsion of the proton is the positive electrostatic potential (shades of blue). Negative regions (red) are mostly located over the electronegative atoms; O and N atoms, which are the most reactive sites for deficient electron species, while most positive regions (blue) are around the H atoms, which are the most reactive sites for a species rich in electrons, according to the MESP and Contour plot for the title compound.
Figure 5: MESP and Contour plot of the title compound.
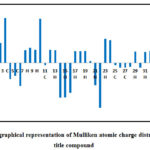
Because of the impact of these charges on dipole moment, polarizability, and electronic characteristics, the estimation of Mulliken nuclear charges plays an important role in quantum compound figurings. To visualise charge distributions over the molecule and identify the electron population of each atom, as detailed in Table 6 and Figure 6. The Mulliken charges are measured at 6-311G(d,p) with B3LYP functional. The atom C3 has the highest positive charge [0.459435 e(M)] in the title compound. This has happened due to its attachment with the oxygen and nitrogen atoms. The next to it is C10 with Mulliken atomic charge value of 0.44955210. The atom with the highest negative charge is N22 [-0.461342e(M)]. Out of five oxygen atoms, O14 has the maximum negative charge[-0.357489e(M)]. All hydrogen atoms are found to be positive in terms of Mulliken nuclear charges.
Figure 6: The graphical representation of Mulliken atomic charge distribution in the title compound.
Ethyl 6-amino-5-cyano-2-methyl-4-(4-nitrophenyl)-4H-pyran-3-carboxylate is synthesized by a three-component one-pot condensation reaction by stirring method at 80℃ using PEG-400-H2Oreaction media and catalytical amount of sulfamic acid. The synthesized compound is characterized by UV, IR, and 1H NMR spectral techniques. DFT-based calculations such as optimized geometry, UV-Visible, and vibrational analysis, and frontier molecular orbital analysis are performed. The theoretical vibrational analysis compared to the observed experimental IR spectrum. The HOMO-LUMO analysis revealed the compound’s stability as well as the transfer of charge within the molecule. Over the electron rich pyran ring, HOMO of the compound is delocalized, while LUMO is delocalized on the 4-nitrophenyl ring. DMSO solvent was found to decline the HOMO-LUMO energy and thereby caused the hypsochromic shift. Molecular electrostatic surface potential investigations show that hydrogen atoms accumulate a positive potential.The most negative and positive atoms in the title compound were disclosed by Mulliken nuclear charges. Total energy, heat capacity, and entropy parameters were disclosed in thermochemical analysis. We believed that this study would be beneficial to the researchers working in the theoretical and heterocyclic chemistry.
Acknowledgement
The authors are thankful to former Professor Dr. A.B. Sawant for his generous help in the Gaussian study. Dr. Aapoorva P. Hiray Coordinator, M. G. Vidyamandir institute is gratefully acknowledged for the Gaussian package.
Conflict of Interest
No potential conflict of interest was reported by the authors
References
Goel, A. and Ram, V.J., 2009. Natural and synthetic 2H-pyran-2-ones and their versatility in organic synthesis. Tetrahedron, 65(38), pp.7865-7913. CrossRef
Geen, G.R., Evans, J.M., Vong, A.K., Katritzky, A.R., Rees, C.W. and Scriven, E.F.V., 1996. Pyrans and their benzo derivatives: applications. Comprehensive heterocyclic chemistry II, 5, pp.469-500. CrossRef
Shehab, W.S. and Ghoneim, A.A., 2016. Synthesis and biological activities of some fused pyran derivatives. Arabian Journal of Chemistry, 9, pp.S966-S970. CrossRef
Kumar, D., Sharma, P., Singh, H., Nepali, K., Gupta, G.K., Jain, S.K. and Ntie-Kang, F., 2017. The value of pyrans as anticancer scaffolds in medicinal chemistry. RSC advances, 7(59), pp.36977-36999. CrossRef
Mar’yasov, M.A., Davydova, V.V., Sheverdov, V.P., Nasakin, O.E. and Gein, V.L., 2016. Synthesis and Antimicrobial, Analgesic, Antipyretic, and Immunotropic Activity of Methyl 3-Aryl-6-Amino-4-Aryl-5-Cyano-4H-Pyran-2-Carboxylates. Pharmaceutical Chemistry Journal, 50(8), pp.519-522. CrossRef
Yu, B., Qi, P.P., Shi, X.J., Shan, L.H., Yu, D.Q. and Liu, H.M., 2014. Discovery of novel steroidal pyran–oxindole hybrids as cytotoxic agents. Steroids, 88, pp.44-52. CrossRef
Zhang, T.T., Lu, C.L. and Jiang, J.G., 2015. Antioxidant and anti-tumour evaluation of compounds identified from fruit of AmomumtsaokoCrevostetLemaire. Journal of Functional Foods, 18, pp.423-431. CrossRef
Baker, D., University of Tennessee Research Foundation, 1998. Pyran-chromenone compounds, their synthesis and anti-HIV activity. U.S. Patent 5,843,990.
Gurunanjappa, P., Ningappa, M.B. and Kariyappa, A.K., 2016. Synthesis of pyrazole fused pyran analogues: Antimicrobial, antioxidant and molecular docking studies. Chemical Data Collections, 5, pp.1-11. CrossRef
Safari, F., Hosseini, H., Bayat, M. and Ranjbar, A., 2019. Synthesis and evaluation of antimicrobial activity, cytotoxic and pro-apoptotic effects of novel spiro-4 H-pyran derivatives. RSC advances, 9(43), pp.24843-24851. CrossRef
Adole, V.A., Pawar, T.B., Koli, P.B. and Jagdale, B.S., 2019. Exploration of catalytic performance of nano-La 2 O 3 as an efficient catalyst for dihydropyrimidinone/thione synthesis and gas sensing. Journal of Nanostructure in Chemistry, 9(1), pp.61-76. CrossRef
Abbasi, M., 2017. β‐Cyclodextrin as an Efficient and Recyclable Supramolecular Catalyst for the Synthesis of Heterocyclic Compounds. Journal of the Chinese Chemical Society, 64(8), pp.896-917. CrossRef
Maleki, A., Ghassemi, M. and Firouzi-Haji, R., 2018. Green multicomponent synthesis of four different classes of six-membered N-containing and O-containing heterocycles catalyzed by an efficient chitosan-based magnetic bionanocomposite. Pure and Applied Chemistry, 90(2), pp.387-394. CrossRef
OuldM’hamed, M., 2015. Ball milling for heterocyclic compounds synthesis in green chemistry: a review. Synthetic Communications, 45(22), pp.2511-2528. CrossRef
Adole, V.A., 2021. Synthesis, Antibacterial, Antifungal and DFT Studies on Structural, Electronic and Chemical Reactivity of (E)-7-((1H-Indol-3-yl) methylene)-1, 2, 6, 7-tetrahydro-8H-indeno [5, 4-b] furan-8-one. Advanced Journal of Chemistry-Section A, 4(3), pp.175-187.
Bihani, M., Bora, P.P., Bez, G. and Askari, H., 2013. Amberlyst A21: A reusable solid catalyst for green synthesis of pyran annulated heterocycles at room temperature. ComptesRendusChimie, 16(5), pp.419-426. CrossRef
Lian, X.Z., Huang, Y., Li, Y.Q. and Zheng, W.J., 2008. A green synthesis of tetrahydrobenzo [b] pyran derivatives through three-component condensation using N-methylimidazole as organocatalyst. MonatsheftefürChemie-Chemical Monthly, 139(2), pp.129-131. CrossRef
Maleki, A. and Hajizadeh, Z., 2019. Magnetic AluminosilicateNanoclay: a Natural and Efficient Nanocatalyst for the Green Synthesis of 4 H-Pyran Derivatives. Silicon, 11(6), pp.2789-2798. CrossRef
Tabassum, S., Govindaraju, S. and Pasha, M.A., 2015. Ultrasound mediated, iodine catalyzed green synthesis of novel 2-amino-3-cyano-4H-pyran derivatives. Ultrasonics sonochemistry, 24, pp.1-7. CrossRef
Chen, L., Lin, C., Lan, Y., Li, Z., Huang, D., Yang, W. and Li, Y., 2020. Visible light-induced green synthesis of 2-amino-4H-chromenes. Environmental Chemistry Letters, 18(6), pp.2157-2163. CrossRef
Adole, V.A., 2021. Computational Chemistry Approach for the Investigation of Structural, Electronic, Chemical and Quantum Chemical Facets of Twelve Biginelli Adducts. Journal of Applied Organometallic Chemistry, 1(1), pp.29-40.
Adole, V.A., Waghchaure, R.H., Pathade, S.S., Patil, M.R., Pawar, T.B. and Jagdale, B.S., 2020. Solvent-free grindstone synthesis of four new (E)-7-(arylidene)-indanones and their structural, spectroscopic and quantum chemical study: a comprehensive theoretical and experimental exploration. Molecular Simulation, 46(14), pp.1045-1054. CrossRef
Elzupir, A.O., Ali, M.K.M., Hussein, R.K., Ibrahem, M.A., Al-Muhanna, M.K. and Ibnaouf, K.H., 2019. Molecular structure, frontier molecular orbital and spectral analysis of dimethylamino chalcones efficient lasing dyes. Journal of Molecular Structure, 1178, pp.285-289. CrossRef
Adole, V.A., Pawar, T.B. and Jagdale, B.S., 2021. DFT computational insights into structural, electronic and spectroscopic parameters of 2-(2-Hydrazineyl) thiazole derivatives: a concise theoretical and experimental approach. Journal of Sulfur Chemistry, 42(2), pp.131-148. CrossRef
Zając, A., Dymińska, L., Lorenc, J., Ptak, M. and Hanuza, J., 2018. Syntheses, spectroscopic properties and molecular structure of silver phytate complexes-IR, UV-VIS studies and DFT calculations. Journal of Molecular Structure, 1156, pp.483-491. CrossRef
Shinde, R.A., Adole, V.A., Jagdale, B.S. and Desale, B.S., 2021. Synthesis, antibacterial and computational studies of Halo Chalcone hybrids from 1-(2, 3-Dihydrobenzo [b][1, 4] dioxin-6-yl) ethan-1-one. Journal of the Indian Chemical Society, 98(4), p.100051. CrossRef
Pathade, S.S., Adole, V.A., Jagdale, B.S. and Pawar, T.B., 2020. Molecular structure, electronic, chemical and spectroscopic (UV-visible and IR) studies of 5-(4-chlorophenyl)-3-(3,4-dimethoxyphenyl)-1-phenyl-4, 5-dihydro-1H-pyrazole: combined DFT and experimental exploration. Material Science Research India, 17(specialissue2020), pp.27-40.
Sadgir, N.V., Dhonnar, S.L., Jagdale, B., Waghmare, B. and Sadgir, C., 2020. Synthesis, Spectroscopic Characterization, Quantum Chemical Study and Antimicrobial Study of (2e)-3-(2, 6-Dichlorophenyl)-1-(4-Fluoro)-Prop-2-En-1-One. Material Science Research India, 17(3), pp.281-293. CrossRef
Waghchaure, R.H. and Pawar, T.B., 2020. Synthesis, characterization, molecular structure, and HOMO-LUMO study of 2-phenylquinoxaline: a DFT exploration. World Journal of Pharmaceutical Research, 9(6), pp.1867-1881.
Pathade, S. and Jagdale, B., 2020. Experimental and Computational Investigations on the Molecular Structure, Vibrational Spectra, Electronic Properties, FMO and MEP Analyses of 4, 6-Bis (4-Fluorophenyl)-5, 6-dihydropyrimidin-2 (1H)-one: A DFT Insight. Physical Chemistry Research, 8(4), pp.671-687.
Halim, S.A. and Ibrahim, M.A., 2021. Synthesis, DFT computational insights on structural, optical, photoelectrical characterizations and spectroscopic parameters of the novel (2E)-3-(4-methoxy-5-oxo-5H-furo [3,2-g] chromen-6-yl) acrylonitrile (MOFCA). Journal of Molecular Structure, 1223, p.129316. CrossRef
Halim, S.A. and Ibrahim, M.A., 2017. Synthesis, DFT calculations, electronic structure, electronic absorption spectra, natural bond orbital (NBO) and nonlinear optical (NLO) analysis of the novel 5-methyl-8H-benzo [h] chromeno [2, 3-b][1, 6] naphthyridine-6 (5H), 8-dione (MBCND). Journal of Molecular Structure, 1130, pp.543-558.’ CrossRef
Frisch, M.J., Trucks, G.W., Schlegel, H.B. and Scuseria, G.E., 2004. etc. Gaussian 03, Revision E. 01.
Beck, Axel D. “Density-functional thermochemistry. III. The role of exact exchange.” The Journal of Chemical Physics 98, no. 7 (1993): 5648-5652. CrossRef
Becke, A.D., 1993. A new mixing of Hartree–Fock and local density‐functional theories. The Journal of chemical physics, 98(2), pp.1372-1377. CrossRef
Millam, J., A.B. Nielsen, A.J. Holder, and J. Hiscocks. “Gaussview, 4.1. 2; Gaussian. Inc.: Wallingford, CT, USA: 2004.”

 , Tejendra A. Rajput2 and Bapu S. Jagdale3
, Tejendra A. Rajput2 and Bapu S. Jagdale3